Search API
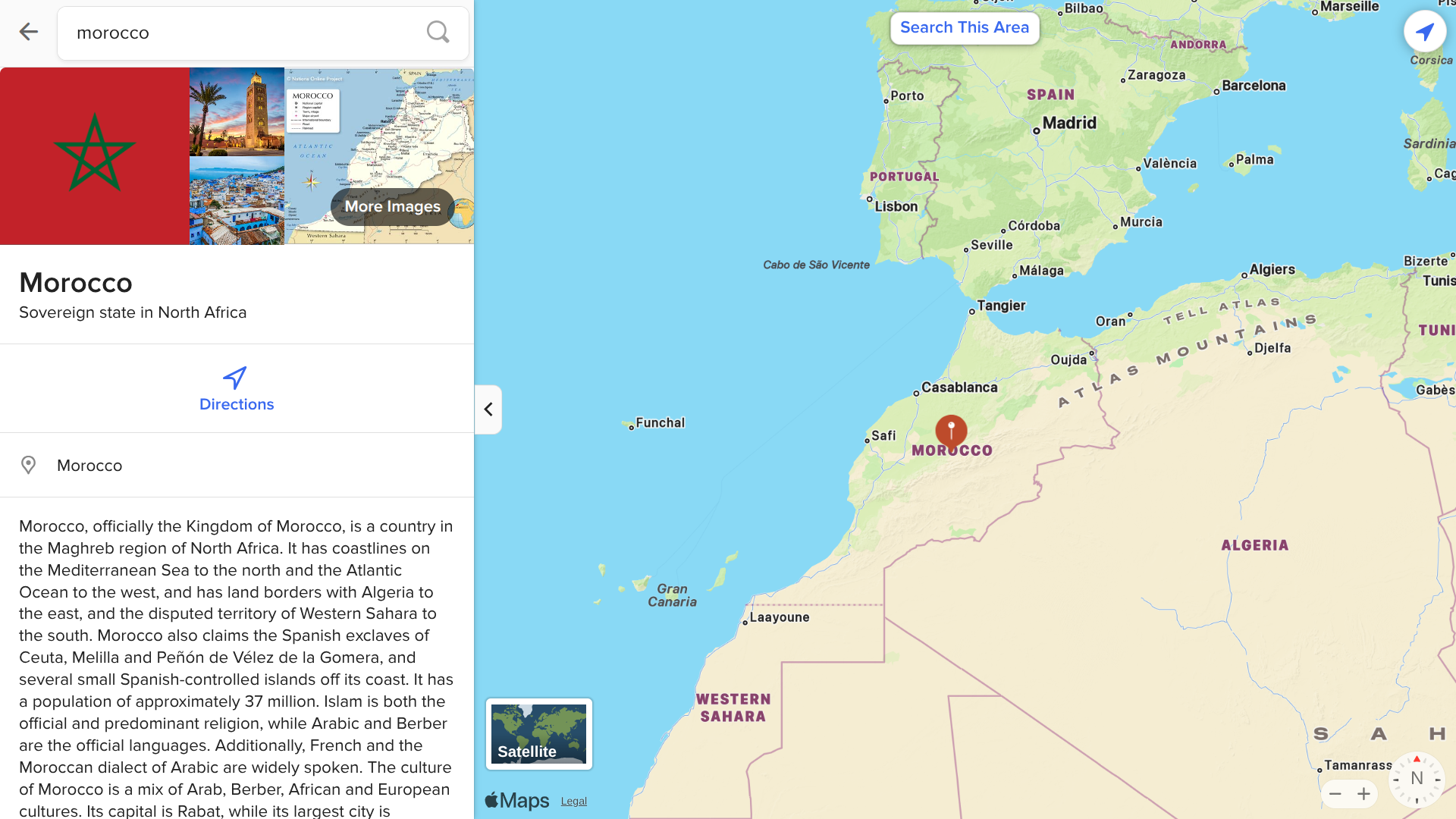
The UK Health Security Agency (UKHSA) today announced that a woman from Barnsley, South Yorkshire, diagnosed at Barnsley Hospital, has died after becoming infected with rabies, following contact with a stray dog during a visit to the Kingdom of Morocco, located in northern Africa.
Currently, the UK's list of rabies-risk areas does not include Monaco.
The UKHSA stated on June 18, 2025, that there is no risk to the broader public about this case, as there is no documented evidence of rabies transmission between people in England. Rabies virus is transmitted through bites and scratches from an infected animal.
Once infected, rabies is nearly always fatal.
Furthermore, rabies does not circulate in either wild or domestic animals in the United Kingdom; however, some species of bats can carry a rabies-like virus, as is the case in the United States.
Dr. Katherine Russell, Head of Emerging Infections and Zoonoses, at UKHSA, commented in a press release, "If you are bitten, scratched or licked by an animal in a country where rabies is found then you should wash the wound or site of exposure with plenty of soap and water and seek medical advice without delay to get post-exposure treatment to prevent rabies."
When administered promptly after exposure, a course of rabies post-exposure treatment is highly effective in preventing the disease. If such an exposure occurs abroad, the traveller should also consult their doctor on return.
The UKHSA and the U.S. CDC suggest that international travelers speak with a travel vaccine consultant regarding rabies immunization options.
As of 2025, the WHO has pre-qualified human rabies vaccines, including Bavarian Nordic's RabAvert vaccine, which is commercially available in the United States.
GSK plc recently announced that it has licensed its Shigella vaccine candidate, altSonflex1-2-3, to India-based Bharat Biotech International Limited (BBIL).
This agreement paves the way for the ongoing development and potential distribution of the vaccine in low- and middle-income countries where Shigella, an acute human infection of the large intestine, is the leading bacterial cause of diarrhoea, posing a significant health threat to children under five.
The urgent challenge posed by increasing antimicrobial-resistant enteric bacteria, including Shigella, highlights the broader impact a vaccine could have beyond helping to reduce illness and mortality rates.
If approved, a Shigella vaccine has the potential to indirectly reduce antibiotic consumption and help combat the rise of antimicrobial resistance.
As of June 18, 2025, there are no U.S. Food and Drug Administration (FDA)- approved Shigella vaccines.
Thomas Breuer, Chief Global Health Officer at GSK, stated in a press release, "With young children in lower-income countries disproportionately impacted by Shigella, the development of a low-cost vaccine is an important goal for global public health."
Following the technology transfer, GSK will collaborate with BBIL on its design of the Phase 3 trial and support BBIL's efforts to secure external funding. This collaboration builds on GSK's existing relationship with the Indian Biotech company, following a product transfer and license agreement in 2021 for the world's first malaria vaccine, RTS,S (Mosquirix™).
Currently, Valneva SE and LimmaTech Biologics AG are co-developing the Shigella4V2 second-generation tetravalent bioconjugate vaccine candidate against shigelllosis, which is progressing in clinical trials.
Local media reported that Bahamian Health Minister Michel Darville announced that The Bahamas is preparing to cancel its contracts with Cuban health professionals.
According to Reuters on June 16, 2025, the reason for the cancellation is that negotiations are underway with the US government.
The NGO Archivo Cuba has reported that Cuban specialist medical advisors in the Bahamas were paid $12,000 per month, while biomedical engineers received $$5,000.
As of June 16, 2025, the U.S. CDC says 'check the vaccines, such as measles or typhoid, and medicines list, and visit your doctor at least a month before your trip to The Bahamas.
Previously, the U.S. and Canadian governments issued travel advisories regarding jet ski activities in the Bahamas.
France's Chikungunya outbreak, centered in the Indian Ocean, may be connected to the recent reporting of locally acquired cases adjacent to the Mediterranean coast.
A press release from the Occitanie Regional Health Agency, issued on June 16, 2025, confirmed a locally transmitted Chikungunya case in the Hérault Department.
Since the beginning of May 2025, the Agency has reported 59 travel-related cases in Occitanie.
This Department includes the city of Montpellier, home to about 800,000 people.
The Agency wrote that anyone 'who have visited this town and who have shown signs suggestive of chikungunya since the beginning of May are invited by the ARS to consult their doctor.'
Recently, an indigenous (locally) Chikungunya case was reported in La Crau (Var) on June 11, 2025.
During 2024, over 20 travel-related cases were reported in France, and one local case in Île-de-France (Paris).
If your plans include visiting areas affected by the Chikungunya outbreak in 2025, the French and US health agencies recommend discussing vaccination options with a travel vaccination expert.
Currently, Chikungunya vaccines are approved by various countries and offer a high degree of protection from this mosquito-transmitted disease.
The Holy Father today urged all young people to participate in the great Jubilee event, dedicated specially to them, as it will be a unique and unforgettable week of celebration, spirituality, reconciliation, and sharing.
On June 15, 2025, Pope Leo XIV addressed a large gathering of children, young people, and adults who had come to Rome for the Jubilee of Sport.
This year's Jubilee will take place from July 28 to August 3, 2025, in Rome, Italy.
Following the first Jubilee of 1300, Pope Boniface VIII established the frequency of Jubilee celebrations as every 100 years. Following a plea from the people of Rome to Pope Clement VI, the frequency was reduced to every 50 years.
After the annexation of Rome to the Kingdom of Italy, the Jubilees were resumed in 1875.
According to the European CDC, no infectious disease events of relevance to the EU/EEA were detected during the Jubilee from June 9 to June 13, 2025.
The ECDC states that the probability of EU/EEA citizens contracting communicable diseases during the Jubilee 2025 is low if general preventive measures are implemented.

While Rabies is a rare disease, it remains a severe viral infection once symptoms appear. Thousands of people die each year from Rabies, mainly in Asia and Africa, with 40% of the cases being children.
According to the World Health Organization (WHO), travellers visiting rabies-endemic areas must be aware of the risk and know what to do if they are bitten or scratched.
WHO estimates that dogs are the primary source of human rabies deaths, causing up to 99% of all transmissions.
European travellers have been amongst those affected, including in June 2025, Spain confirmed a rabies case in a traveller who a dog in Ethiopia had bitten.
However, in the United States, bat bites are responsible for the majority of rabies cases in humans.
In March 2025, a bat found in northern Person County, North Carolina, tested positive for Rabies, marking the second confirmed case in Person County this year.
To reduce the risk of Rabies in Texas, the Department of State Health Services conducts the annual Oral Rabies Vaccination Program bait drop for animals. In its 31st year, this program has proven to be an effective defense against the spread of domestic dog and coyote rabies, as well as the Texas gray fox variant.
From a prevention perspective, two types of vaccines protect people against Rabies: nerve tissue and cell culture vaccines.
As of June 17, 2025, rabies vaccination services are offered at travel clinics and pharmacies, but are only recommended for specific travelers.
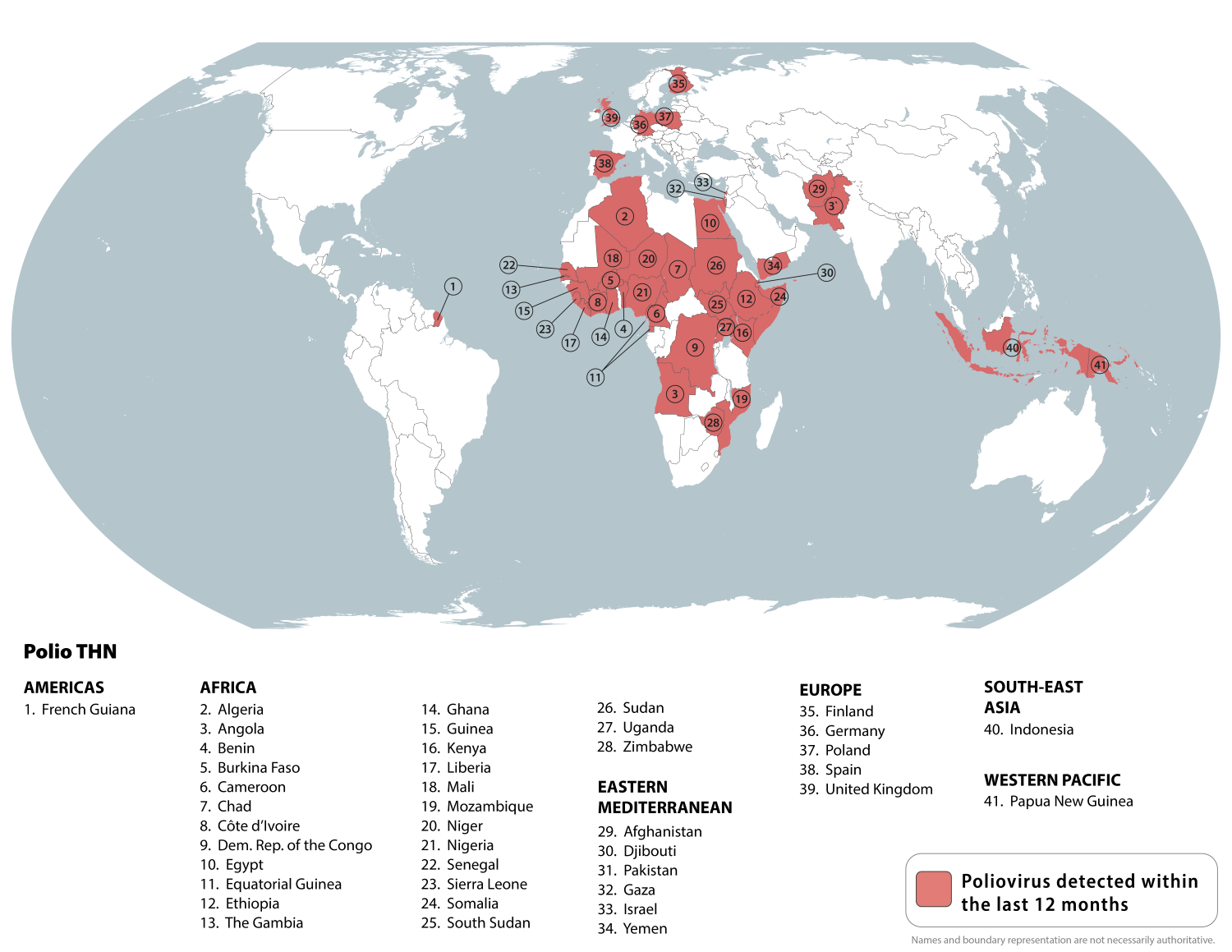
According to today's Travel Health Advisory issued by the U.S. Centers for Disease Control and Prevention (CDC), circulating poliovirus is a risk in 41 countries.
On June 16, 2025, the CDC reissued its Level 2 - Practice Enhanced Precautions alert for countries in Africa, Asia, Europe, the Mediterranean, and the western Pacific.
Last week, the Global Polio Eradication Initiative confirmed six countries had reported cases of wild poliovirus type 1.
The CDC stated that before traveling to any destination listed, including Indonesia, the UK, and Spain, adults who have previously completed the routine polio vaccine series may receive a single, lifetime booster dose of polio vaccine.
Furthermore, children should be fully protected against this severe disease before traveling abroad.
In the United States, the IPV polio vaccine is offered at health clinics and pharmacies.