Search API

In June 2025, the WHO's Dengue Situation Update #723 confirmed that the Western Pacific Region continues to face a high burden of mosquito-borne arboviral diseases, particularly Dengue.
Within the Philippines, Quezon City (QC) declared a Dengue outbreak in February 2025
The QC government today published updated data regarding the ongoing outbreak.
The QC Epidemiology and Surveillance Division (QCESD) dashboard reported on July 1, 2025, that 5,762 dengue cases were reported in 2025, with QC's District 2 confirming the highest number of cases.
The QCESD stated that most fatal dengue cases (23) involved children and young women.
To alert international travelers visiting QC, a metro area with about 3 million residents located north of Manila, the U.S. Centers for Disease Control and Prevention included the Philippines in its Global Dengue Outbreak Advisory issued in June 2025.
Without a preventive vaccine available in the U.S., the CDC recommends that travelers to risk areas should prevent mosquito bites by using an EPA-registered insect repellent, wearing long-sleeved shirts and long pants when outdoors, and sleeping in an air-conditioned room or one with window screens.

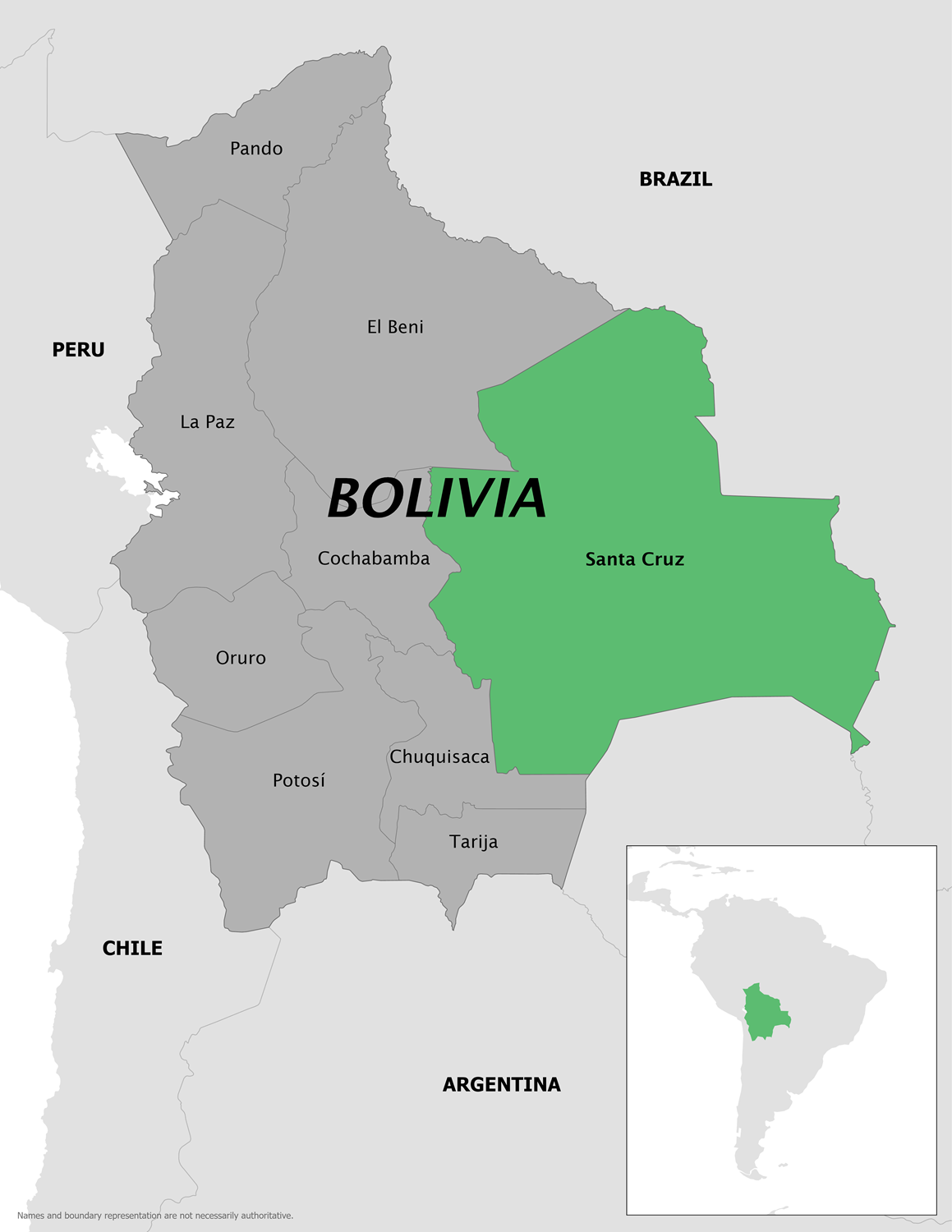
Like many countries in South America, the Plurinational State of Bolivia has been confronted with a multi-year outbreak of Chikungunya.
To alert international travelers visiting Bolivia, the U.S. Centers for Disease Control and Prevention (CDC) issued a Level 2 Travel Health Advisory on June 26, 2025, stating that this year's Chikungunya outbreak is centered in Bolivia's Santa Cruz department.
According to recent data, Bolivia welcomed around 984,000 international tourists in 2024.
Data released at the end of June 2025 indicates Bolivia has reported 3,863 Chikungunya cases this year and 505 cases in 2024.
So far in 2025, about 38 people have returned to the U.S. infected with the Chikungunya virus.
The CDC advises that if you are pregnant, you should reconsider travel to the affected areas, especially if you are nearing the time of delivery. Mothers infected around the time of delivery can pass the virus to their baby before or during delivery.
Newborns infected in this way or by a mosquito bite are at risk for severe illness, including poor long-term outcomes.
Furthermore, vaccination is recommended for most travelers who are visiting an area with a Chikungunya outbreak.
As of June 30, 2025, Chikungunya vaccines are approved for use by the CDC and are commercially available at travel clinics and pharmacies in the United States.
Current polio vaccines are made from either inactivated or weakened versions of the virus. These vaccines have been administered worldwide for many decades.
However, this approach presents challenges in certain situations. It highlights the need for an improved vaccine that does not rely on the virus itself, particularly as the global community strives to end poliovirus infections.
As of June 30, 2025, the U.S. Centers for Disease Control and Prevention identified poliovirus detections in 41 countries.
Developing vaccine formulations that do not use live viruses in their production would be highly beneficial, as it would eliminate the potential safety risks associated with handling and growing the virus, wrote Evaxion A/S in early June 2025.
To address this need, Evaxion received undisclosed funding from the Gates Foundation to help eradicate polio worldwide by exploring design options for a new and innovative vaccine.
This project will combine Evaxion’s leading and clinically validated AI-Immunology™ platform to identify and combine various antigens to combat the virus. Based on these findings, several new antigen constructs will be designed for selection and validation.
“We are thrilled to receive support from the Gates Foundation and help the world achieve the goal of completely eradicating polio. We are excited to apply our AI-Immunology™ platform to combat yet another infectious disease. The grant allows for further application and validation of our platform without adding to our operational spend,” says Christian Kanstrup, CEO of Evaxion, in a press release on June 3, 2025.
The inactivated (killed) polio vaccine (IPV) was developed in 1955 to produce antibodies in the blood that target all three poliovirus types, thereby preventing the spread of the virus, and has been offered in the U.S. since 2000.
IPV vaccinations are offered at clinics and pharmacies in the U.S.

Rabies has been present in the wildlife population in New Jersey since 1989, affecting the entire state.
Rabies is a virus that can affect any mammal, including cats, dogs, raccoons, skunks, and bats, the unfortunate source of most rabies cases in the United States.
In NJ, groundhogs have been a significant source of rabies.
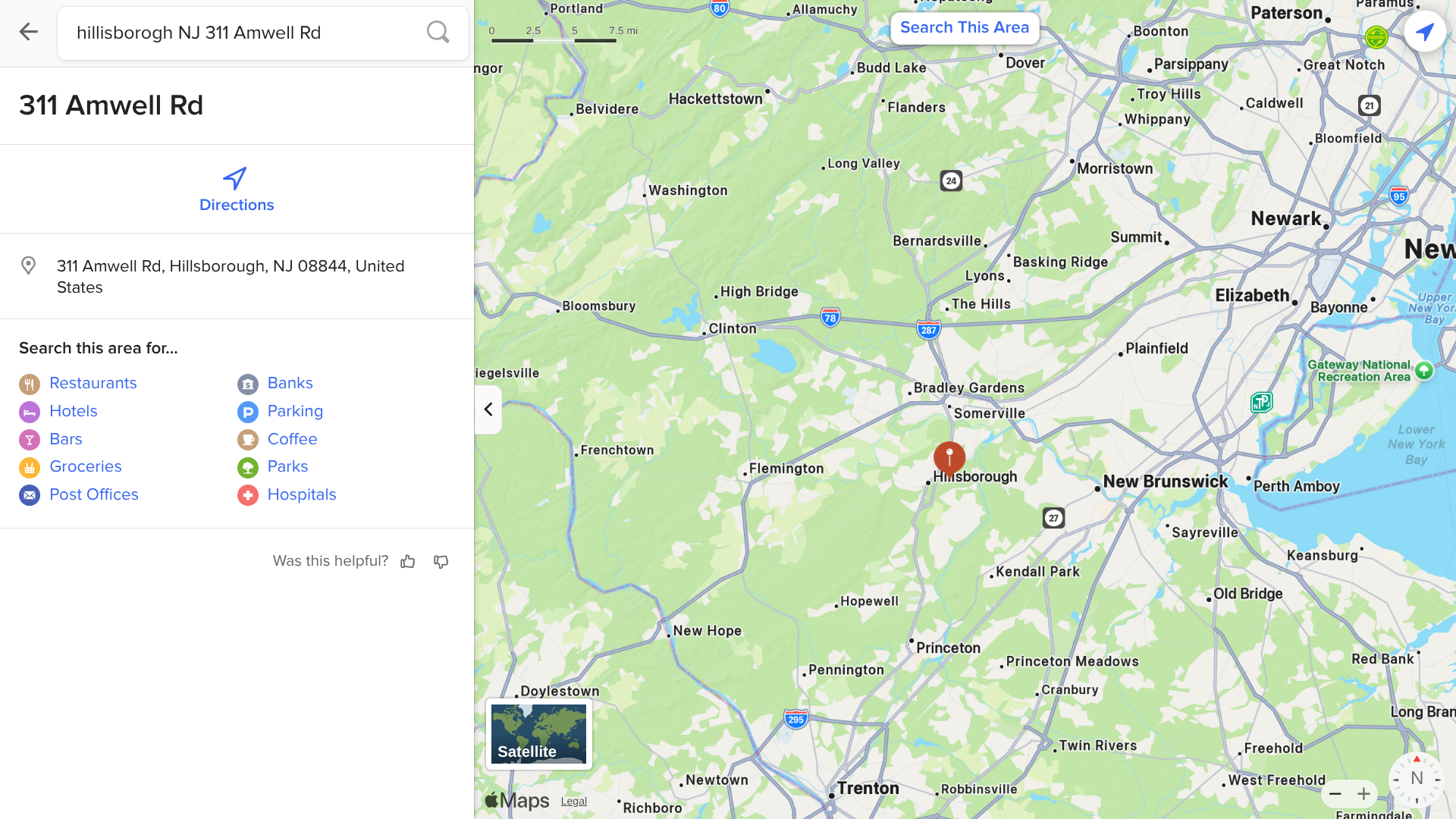
Recently, the Hillsborough Health Department tested a groundhog that bit two people outside of a business at The Landing business complex. This animal tested positive for rabies.
As of June 27, 2205, rabies vaccinations of all cats and dogs, with few exceptions, are required in Hillsborough Township.
The virus lives in the central nervous system fluid and saliva of the affected animal. This infectious fluid must enter the host’s body thost's an opening put there by a bite, or a scratch, or through exposure of an open cut, or rubbing the eyes after touching saliva.
'If any anima' bites you, wash the wound immediately with soap and water and seek medical attention,' says the NJ Hea'th Department.
Additionally, rabies vaccination is immediately recommended for people bitten by a rabid animal.
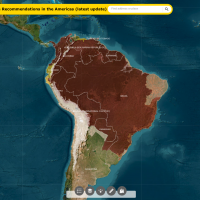
Since 1952, when the first Japanese Encephalitis (JE) case in India, this mosquito-transmitted disease has been a significant public health concern.
Local media reported in late June 2025 that an outbreak of JE is responsible for 32 people being admitted to a hospital, with four related fatalities, in Assam, a state in northeastern India.
Since April 2025, the Japanese Encephalitis virus (JEV) has primarily affected areas with pig-rearing and paddy field activity, both known risk zones for virus transmission by infected mosquitoes.
These mosquitoes acquired the JEV from pigs, birds, and sheep.
According to data from the Integrated Disease Surveillance Programme, about 925 JE cases were detected in Assam in 2024.
To help reduce the impact of this severe disease, JE vaccinations have been started in Assam's nine districts.
Assam is located south of the eastern Himalayas along the Brahmaputra and Barak River valleys, just to the east of Nepal, where twenty-nine districts have reported JE cases.
To alert international travelers visiting Assam and Nepal, the U.S. CDC recommends vaccination for travelers who are moving to an area with an outbreak or spend extended periods in areas with Japanese encephalitis cases.
When departing from the United States, an approved JE vaccine (IXIARO®) is commercially offered at travel clinics and pharmacies.
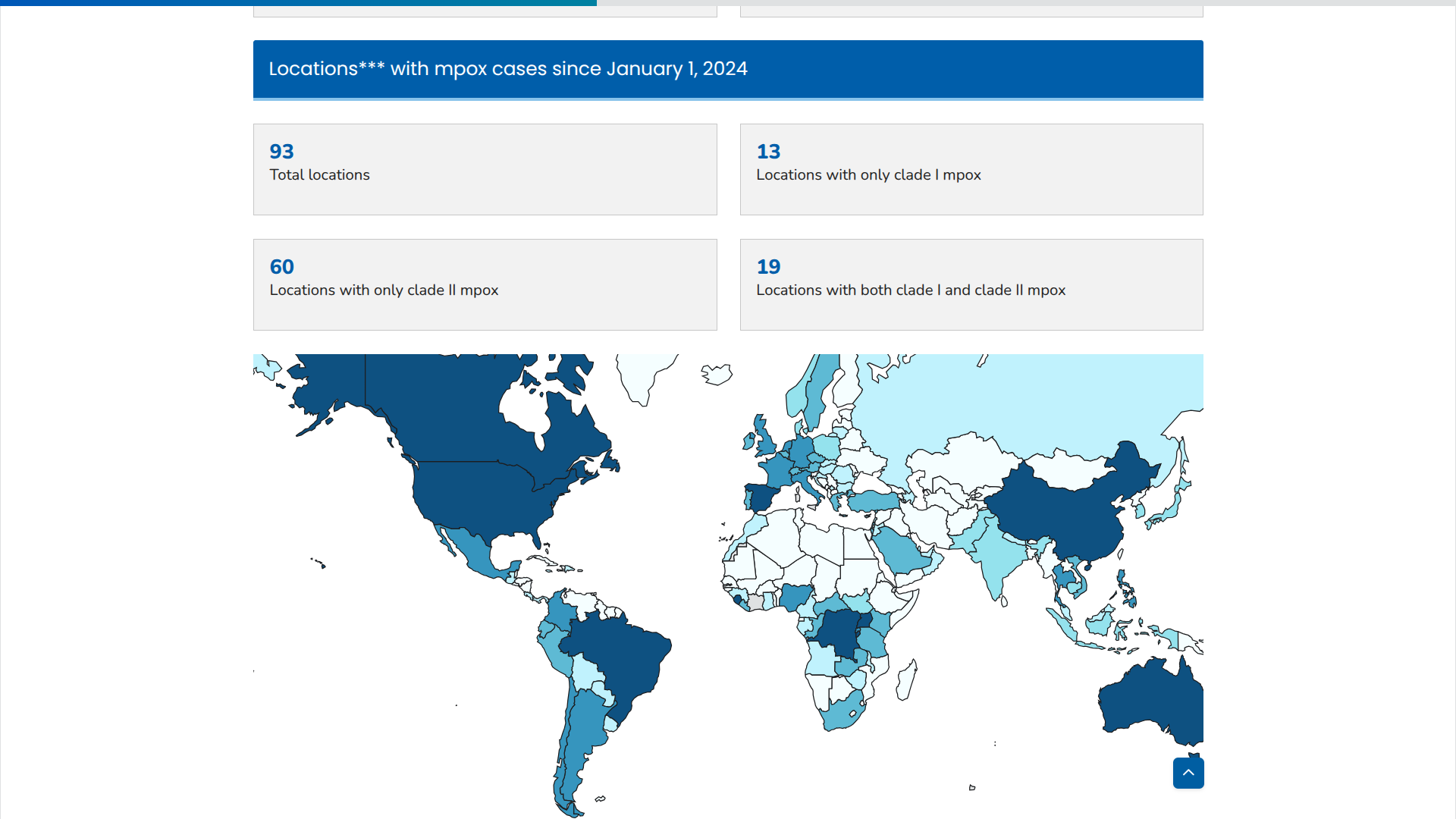
The World Health Organization (WHO) has published the 54th situation report for the multi-country outbreak of mpox, which began in May 2022.
The WHO Director-General recently confirmed that the ongoing upsurge of mpox cases continues to constitute a public health emergency of international concern.
On June 27, 2025, the WHO confirmed that during May 2025, a total of 6,823 confirmed mpox cases and 16 deaths (Case Fatality Ratio: 0.2%) were reported. The majority of mpox instances continue to be reported from the WHO African Region, with 18 countries currently experiencing active ongoing transmission.
Since the last WHO report, Ethiopia and Italy have reported their first cases of mpox, caused by the clade Ib monkeypox virus (MPXV).
In addition, North Macedonia, the Republic of the Congo, and Togo have reported their first cases of mpox clade IIb MPXV. Albania has reported its first mpox case.
According to a disease prevention update, the WHO reported that seven African countries have initiated mpox vaccination. More than 731,000 doses of the MVA-BN (JYNNEOS) vaccine have been administered to date.
In the United States, there have been four reported cases of clade I mpox in people who had recently traveled to affected areas in Central and Eastern Africa.
The U.S. CDC advises that when visiting countries in Africa, such as the Democratic Republic of the Congo, you should get your first mpox vaccine at least 6 weeks before traveling, if possible. After completing your first and second vaccine doses, which are given four weeks apart, it takes about two more weeks to achieve the best protection against mpox.
Whether or not you've been vaccinated, continue to reduce your risk of getting mpox, says the CDC.