Search API
According to an update from the Global Polio Eradication Initiative (GPEI) on June 4, 2025, six countries reported additional polio cases last week.
The Islamic Republic of Pakistan, located in South Asia, reported a case of wild poliovirus type 1 (WPV1) from Gilgit-Baltistan, bringing its total number of cases to 11 in 2025.
Additionally, two WPV1-positive environmental samples were reported from Balochistan and Punjab, indicating the risk of additional cases being reported in 2025.
In 2024, 74 WPV1 cases were reported in Pakistan.
According to the European CDC, the last WPV infection in Europe was in 1998, and the World Health Organization declared the European Region polio-free in 2002.
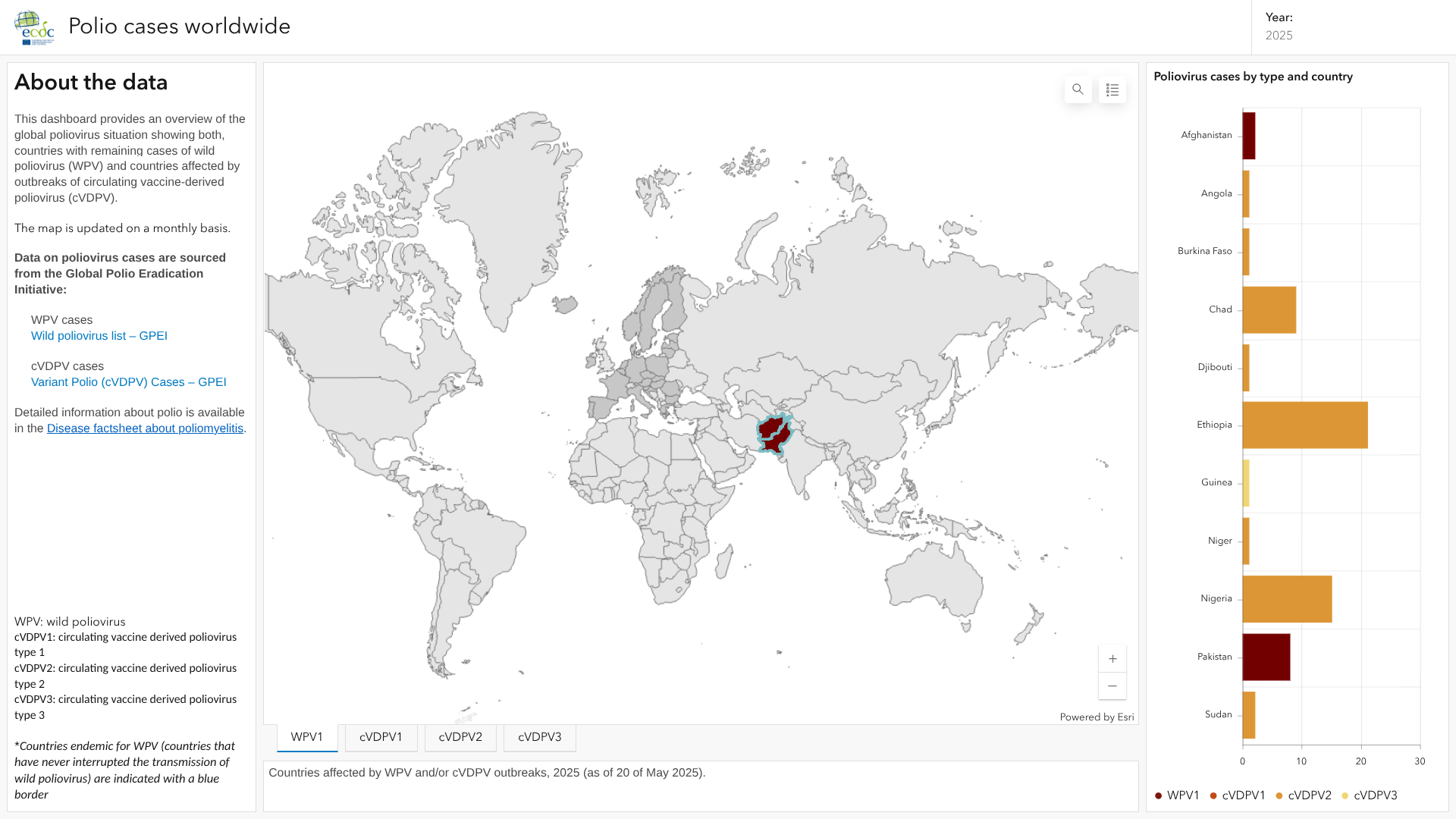
Five other countries reported circulating vaccine-derived poliovirus type 2 (cVDPV2) cases last week: Chad, Angola, Chad, Ethiopia, Niger, and Yemen.
Additionally, Papua New Guinea reported acute flaccid paralysis cases have been reported across 11 provinces, with 20 cases testing negative for poliovirus and 28 cases still under investigation, as of June 6, 2025.
Since polio is a vaccine-preventable disease, the ECDC published a guide for children and adults that focuses on strengthening the capacities of healthcare providers to better address concerns about vaccination and tackle obstacles to vaccination uptake.
In the United States, Polio vaccination has been part of the routine childhood immunization schedule for decades. Since 2000, the Inactivated polio vaccine (IPV) has been the only polio vaccine offered. In July 2022, a case of polio caused by VDPV2 in an unvaccinated individual from Rockland County, New York.
The U.S. CDC recommends that an IPV booster dose may be advisable when visiting a poliovirus endemic area in 2025.
The World Health Organization (WHO) Director-General, Dr Tedros Adhanom Ghebreyesus, today announced that the mpox upsurge continues to meet the criteria of a public health emergency of international concern (PHEIC).
As of June 9, 2025, this PHEIC has been declared, based on the continuing rise in the number of cases, including a recent increase in West Africa, and likely ongoing undetected monkeypox virus (MPXV) transmission in some countries beyond the African continent.
The Director-General also concurred with and issued the Committee's revised temporary recommendations to Member States experiencing mpox outbreaks.
Regarding preventive vaccinations, the WHO advises preparing for and implementing targeted use of vaccines for "Phase 1- Stop the outbreak" through the identification of disease hotspots and targeting those groups at high risk of mpox exposure to interrupt sustained community transmission.
As of early June 2025, the U.S. CDC states that JYNNEOS is a two-dose vaccine developed to protect against mpox and smallpox. People need to receive both doses of the vaccine for optimal protection against mpox.
In the United States, JYNNEOS® is commercially offered at health clinics and pharmacies, with insurance options available.
Furthermore, to be most effective, mpox vaccination should be included as part of broader prevention activities and routine sexual health care, such as HIV or gonorrhea.
'Whether or not you've been vaccinated, continue to reduce your risk of getting mpox,' writes the CDC.
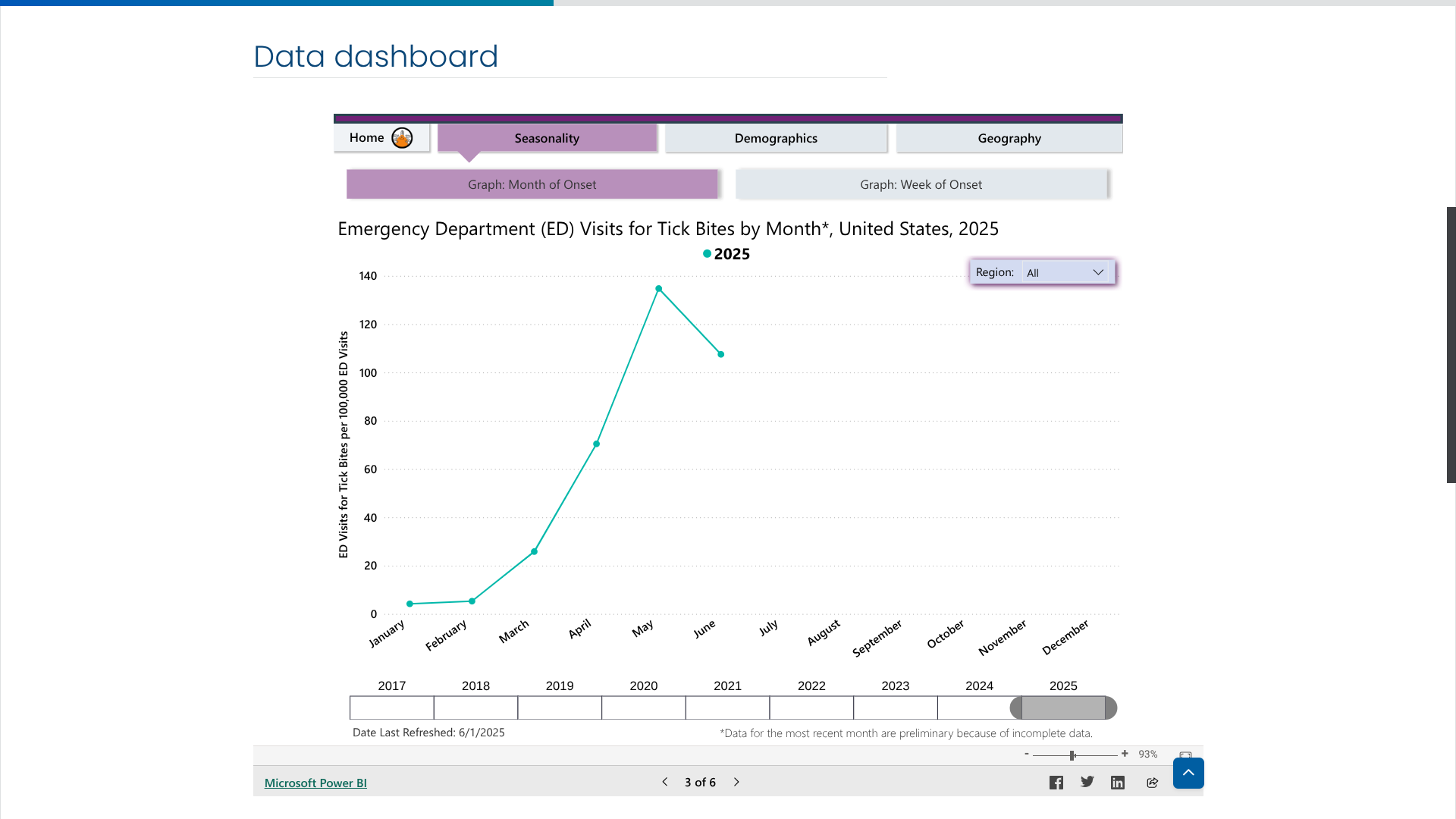
While not every tick bite can pass the Lyme disease virus to a person, avoiding bites is the best way to prevent this severe disease in 2025. Since tick bites can occur year-round, they generally peak during the warmer months in the northeastern United States, between April and September.
However, this year has started differently.
According to new data posted by the U.S. CDC's Tick Bite Data Tracker, the number of emergency room visits related to a tick bite has been decreasing over the past month.
The CDC's data may not accurately reflect the actual spread of Lyme disease into the Midwest in 2025.
Last year, the Michigan Department of Health and Human Services confirmed Lyme disease was the most common tick-borne disease in Michigan. Cases in Michigan have increased by 168% over the last five years.
As a Lyme disease prevention update, the U.S. Food and Drug Administration has not yet approved a vaccine.
But an innovative Lyme disease vaccine candidate (VLA15) is progressing in late-stage clinical trials.
According to data released by the Ministry of Health & Family Welfare and Department of Health & Family Welfare in India, Kerala remains the most affected state during the resurgence of COVID-19 in 2025.
As of June 8, 2025, COVID-19 cases have also been reported in Gujarat, West Bengal, and Delhi.
Local media reported 358 new infections, bringing India's active case tally above the 6,400 milestone this year. Since January 2025, about 65 COVID-19-related deaths have been reported in India.
The Ministry posted on X today, 'India demonstrated remarkable solidarity by sending over 30 crore vaccine doses to 101 countries. This means every second country received a Made-in-India COVID vaccine, showcasing India's commitment to global health and vaccine equity.'
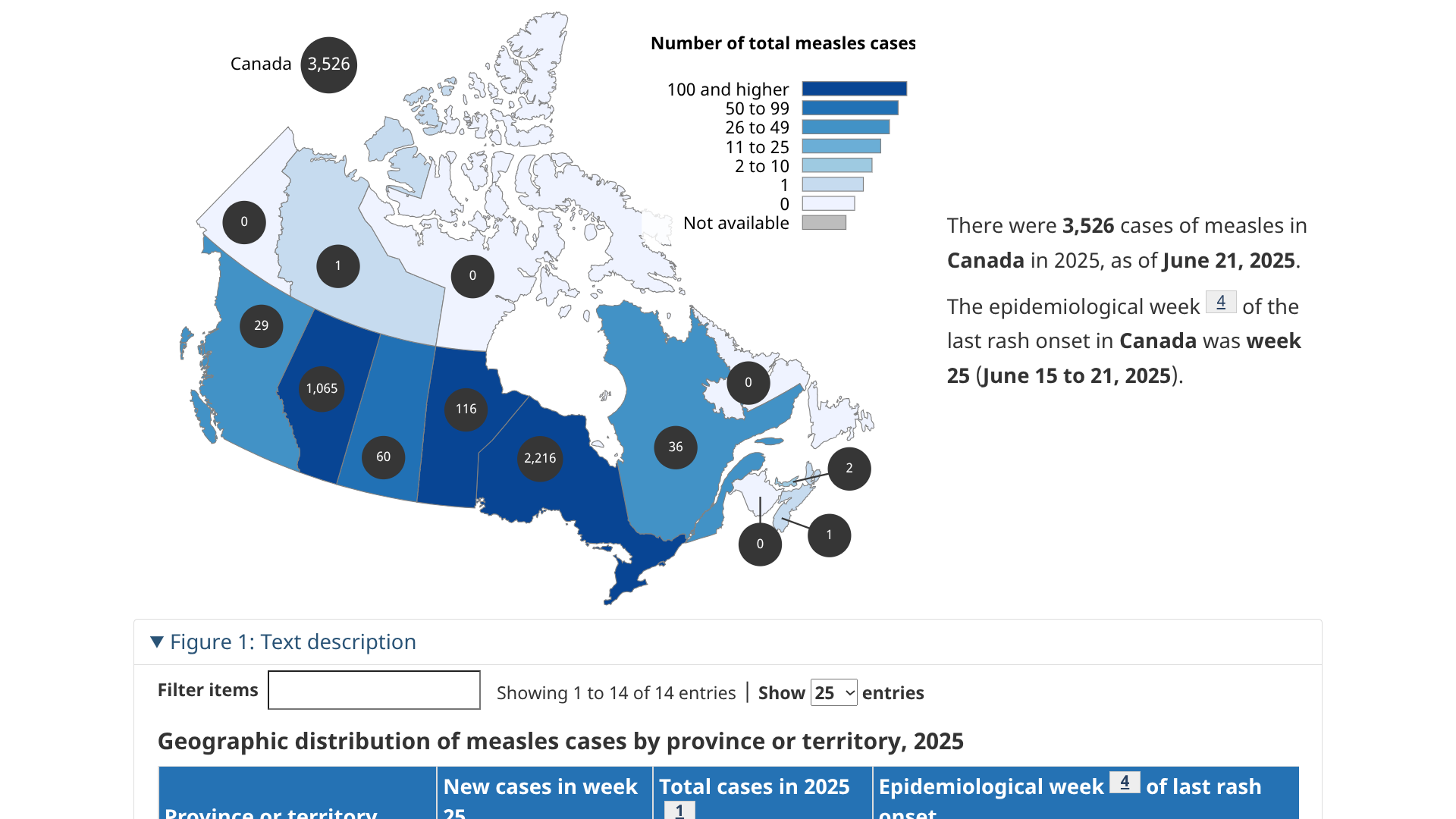
A multijurisdictional measles outbreak has occurred in Canada, beginning in New Brunswick in October 2024. This year, it has spread in Ontario (1,949), and related cases have been reported in Alberta, British Columbia, Manitoba, the Northwest Territories, Nova Scotia, Prince Edward Island, Quebec, and Saskatchewan.
Canada Health's Measles and Rubella Weekly Monitoring Report states that, of the 2,755 measles cases (2,429 confirmed, 326 probable) reported in 2025, 1,867 cases are linked to this outbreak as of June 2, 2025.
At the end of May 2025, only 244 new measles cases were reported in one week, indicating a continued decline of this outbreak.
The government says, 'If you're infected with the measles virus, you can spread it to others. This is possible from 4 days before the onset of the rash to 4 days after the rash begins. Isolate at home and call a health care provider immediately. They will advise you on what to do.'
As of June 8, 2025, measles vaccination services are offered throughout Canada.
While about half of the world’s population is at risk of malaria, the African Region accounts for about 95% of all cases.
However, this mosquito-transmitted disease has been detected in various unusual locations in 2025.
In the Western Pacific region, which includes Australia, Papua New Guinea (PNG), New Zealand, Vanuatu, and the Solomon Islands, malaria cases have been reported.
Malaria in Australia is commonly recorded in returned international travellers, with about 100 imported cases recorded in Queensland each year. But so have locally acquired cases.
For example, Queensland Health recently reported a second locally acquired malaria case in a Torres Strait LGA resident.
As of June 2, 2025, investigations into this malaria case are ongoing.
In 2025, 71 malaria notifications were reported, of which 97% were related to international travel, predominantly with PNG and the Solomon Islands.
The last locally acquired malaria outbreak in the Torres Strait was in 2023, with five cases detected in northern Torres Strait Islands.
From a disease prevention perspective, neither malaria vaccine is offered in Australia as of June 6, 2025.

According to a Case Report published by the Journal of Infection and Chemotherapy (Volume 31) in June 2025, Japanese encephalitis (JE) has emerged in a previously non-reported area in Japan, suggesting that the number of JE patients may be underestimated in Japan.
This report describes three cases of JE in a single hospital in Narita over three years.
These researchers suggest that physicians in Japan should consider JE as a differential diagnosis in 2025 when encountering cases of encephalitis or meningitis with unknown etiology during the warm season, even in areas where JE has not been previously reported.
This prefecture is located a few miles east of Tokyo, a city with over 35 million residents.
JE is an infection of the brain caused by the Japanese encephalitis virus, which is spread to people through the bite of an infected mosquito or close contact with livestock, such as birds, goats, and pigs.
In Japan, JE used to be endemic, and more than 1000 JE patients had been reported before the 1960s.
However, the introduction of the JE vaccine in 1954 and its widespread use in childhood vaccination from 1967 dramatically reduced the disease burden. As of 2022, only one case had been reported in Chiba and Ibaraki prefectures, respectively, in the past 10 years, and both cases occurred in areas famous for pig farming, far from Narita City.
As of June 5, 2025, the U.S. CDC recommends JE vaccination for specific people visiting Japan.
The CDC recommends vaccination for travelers who are moving to an area with JE, spend long periods in areas with JE present, or frequently travel to those areas in Japan.
JE vaccination is not recommended for travelers planning short-term trips to urban areas or those traveling to places with no clear JE season.
When departing for Japan or any other JE outbreak area, such as Australia, Valneva SE's IXIARO® (JESPECT®) vaccine is commercially offered at travel clinics and pharmacies in 2025.