Search API
The Cook Islands in the South Pacific Ocean recently confirmed it is also experiencing a dengue fever outbreak in May 2025.
On May 22, 2025, the Ministry of Health officially declared a dengue fever outbreak in Rarotonga, a city with a population of 10,000, following the confirmation of seven cases over the past 18 days.
Since February, a total of 11 isolated cases have been reported.
The Ministry confirmed there are no dengue cases in the Pa Enua.
Bob Williams, Secretary of Health, commented in a media release, “We urge everyone to help stop the spread of dengue in our communities. By working together, we can protect our families and prevent dengue from reaching the Pa Enua."
"Let’s take action now.”
The Cook Islands Government has recently announced $4.1 million in funding to bring Pa Enua residents to Rarotonga in July for the 2025 Te Maeva Nui celebrations, which will take place from July 25 to August 5 this year.
All clinics and health facilities in the Cook Islands remain on alert and are well-equipped to manage any further cases, says the Ministry.
The World Health Organization and regional health partners have been informed, and no travel restrictions have been issued; however, travelers are advised to take precautions.
When the U.S. CDC updated its Level 1 - Practice Usual Precautions, Dengue Travel Health Advisory on May 22, 2025, it did not list the Cook Islands. The CDC identified Fiji, French Polynesia, and the Philippines.
The CDC recommends several routine and travel vaccines for visitors to the Cook Islands, but not the new dengue vaccine.

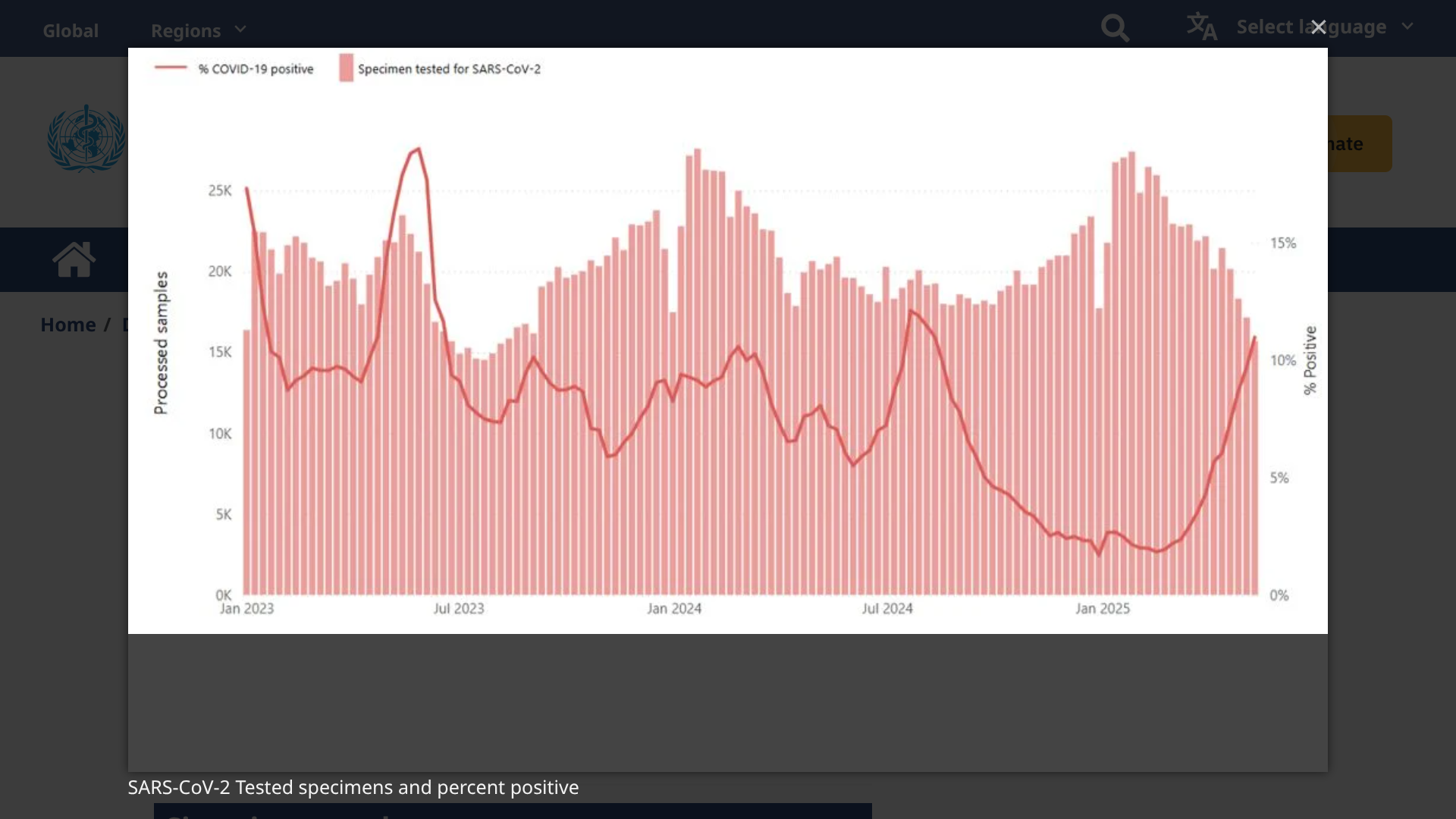
Since early 2025, SARS-CoV-2 virus activity has been increasing globally, with the positivity rate reaching 11% in some countries. However, unlike in past years, activity is now very regional.
According to the World Health Organization (DON572) on May 28, 2025, this respiratory disease increase is primarily observed in countries in the Eastern Mediterranean, South-East Asia, and Western Pacific regions.
This DON says recent increases in SARS-CoV-2 activity are broadly consistent with levels observed during the same period last year. But virus surveillance has been limited in 2025
Countries in the African Region, European Region, and the Region of the Americas are currently reporting low levels of SARS-CoV-2 activity with percent positivity from sentinel or systematic virological surveillance sites ranging from 2% to 3%.
WHO advises all Member States to continue applying a risk-based, integrated approach to managing COVID-19, including vaccinations. Currently approved COVID-19 vaccines continue to protect against severe disease and death.
The WHO and its Technical Advisory Group on COVID-19 Vaccine Composition (TAG-CO-VAC) continue to regularly assess the impact of variants on the performance of COVID-19 vaccines to inform decisions on updates to vaccine composition.
In the latest recommendation published in mid-May 2025, the WHO TAG-COVAC advised that monovalent JN.1 or KP.2 remain appropriate COVID-19 vaccine antigens; monovalent LP.8.1 is a suitable alternative vaccine antigen.
An estimated 39.2 million individuals, across 90 reporting Member States, had received a dose in 2024. Uptake was notably higher among older adults, with coverage reaching 5.1% in the European Region and 3.6% in the Region of the Americas, compared to less than 0.5% in other regions.
In the United States, the COVID-19 vaccination policy was updated in May 2025.
Amidst the peaceful waves of the South Pacific, the Republic of Vanuatu's 330,000 residents have steadily recovered from a devastating earthquake.
Since December 2024, the Infrastructure and roads in Port Vila and the surrounding area have been updated.
In response to this effort, the U.S. Department of State updated its Travel Advisory for Vanuatu to reflect a lowering from Level 3 to Level 1 due to the removal of the Natural Disaster indicator.
As of May 22, 2025, the State Department advises visitors to Vanuatu to exercise normal precautions.
The Director of Tourism recently informed local media that the total number of air arrivals in January 2025 was 9,353. Among visitor arrivals, Australian visitors accounted for the highest proportion at 59%.
And when visiting this island country in Melanesia, northeast of Australia, enroll in the Smart Traveler Enrollment Program to receive digital alerts and make it easier to locate you in an emergency.
Unfortunately, the State Department advises reconsidering travel to Papua New Guinea due to civil unrest, where the local U.S. Embassy is located.
From a health perspective, the U.S. CDC suggests several routine and travel vaccines to prevent diseases when visiting Vanuatu in May 2025.
For example, the CDC suggests the typhoid vaccine.
While malaria is present in Vanuatu, vaccination is not recommended.
The CDC recommends that travelers visiting Vanuatu take prescription medication to prevent malaria. Depending on the medication you take, you will need to start taking it multiple days before your trip, as well as during and after your trip.
In April 2025, the Ministry of Health reaffirmed its ongoing commitment to protecting the health and well-being of all people in Vanuatu through safe, effective, and evidence-based vaccination programs.
'Immunization remains one of the most powerful and proven tools to prevent a wide range of infectious diseases and promote public health,' wrote the Ministry.
These medicines and vaccines are available in the U.S. at travel clinics and pharmacies as of May 2025.

Since the end of the recent pandemic, medical care has become a global industry. Research reveals that about 1.4 billion passengers are expected to fly in 2025.
The Medical Value Travel (MVT) population, which travels to countries seeking healthcare services, is experiencing rapid growth worldwide.
According to the Federation of Indian Chambers of Commerce (FICCI), the global MVT market size was valued at $115.6 billion in 2022 and is expected to reach approximately $286.1 billion by 2030.
In India, the MVT is expected to grow at a CAGR of 21.1% from 2020 to 2027 and is projected to reach $13 billion by 2026.
Bangladesh, Iraq, Maldives, Afghanistan, Oman, Yemen, Sudan, Kenya, Nigeria, and Tanzania account for about 88% of the total international patients visiting India.
The leading health services offered in India are for heart surgery, knee transplant, cosmetic surgery and dental care as the cost of treatment in India is considered to be the lowest in Asia.
The FICCI video stated on May 26, 2025, 'Realizing the increasing importance of global medical value travel and its significance to the Indian medical industry.'
Recent research indicates that tens of millions of people are not adequately vaccinated before visiting disease-endemic countries.
For example, in Europe, over 4% of returning international travelers who recently displayed symptoms may be infected with a mosquito-transmitted disease, such as chikungunya, dengue, or Zika.
When departing abroad in 2025, the U.S. CDC recommends that travelers consult with a travel vaccine expert about immunization options at least one month before departure.

ImmunityBio, Inc., today announced the signing of a strategic Memorandum of Understanding (MOU) with the Ministry of Investment of Saudi Arabia (MISA), King Faisal Specialist Hospital & Research Centre (KFSHRC), and King Abdullah International Medical Research Center (KAIMRC).
As of May 27, 2025, this multi-party collaboration will introduce the FDA-approved Cancer BioShield platform to Saudi Arabia and the broader Middle East, marking a new era of immune-restorative therapies for cancer patients.
Dr. Patrick Soon-Shiong, Founder, Executive Chairman, and Global Chief Medical and Scientific Officer of ImmunityBio, stated in a press release, “We are honored to work with KAIMRC, KFSHRC, and MISA to bring this transformative technology to the region."
"The BioShield platform changes the way we think about treating cancer, not by destroying the immune system but by restoring and activating it."
"The root cause of early mortality is the collapse of the immune system—lymphopenia is the disease, and cancer is a symptom."
"Together, by considering this a paradigm change, we can build a regional center of excellence for next-generation immunotherapies in which we activate the body’s natural defenses.”
The BioShield platform, powered by Anktiva (nogapendekin alfa inbakicept)—the world’s first FDA-approved IL-15 superagonist to stimulate the proliferation of NK and T cells (lymphocytes)—represents a paradigm shift in cancer care.
Unlike conventional treatments, such as chemotherapy and radiation, which kill and suppress natural killer immune cells, thereby paradoxically catalyzing further spread, BioShield protects and activates the immune system’s natural killer cells and T cells to restore immune function and prolong life.
For the first time in medicine, physicians can address the long-overlooked impact of lymphopenia (loss of NK and T cells), induced by current standards of care of chemotherapy, radiation, or by the cancer itself.
The BioShield is the first therapy in history to specifically address the protection and restoration of lymphocytes, represented by NK, CD8, and CD4 T cells—the most important cells in the body needed to fight cancer and infection.
Treating lymphopenia is an answer to premature death from life-threatening diseases such as cancer and sepsis, and, potentially, to aging and longevity in health.
As of May 2025, Anktiva plus BCG Vaccine is available at various clinical sites in the U.S.


According to a recent blood donor study, about 37% of the residents of the Republic of Paraguay may be infected with the Chikungunya virus.
A research article published in the journal Medical Virology on May 13, 2025, stated that serum samples from 546 blood donors across seven regional districts and Asunción were collected from March to May 2023.
Anti-CHIKV IgG prevalence was 37.2%, with men showing a seroprevalence nearly 10% higher than women, but no significant age-related differences were observed.
Regional variation in CHIKV seroprevalence was not substantial.
In conclusion, this study suggests a high seroprevalence of CHIKV in Paraguayan blood donors, with the notable CHIKV prevalence underscoring the effects of recent outbreaks.
As of May 26, 2025, the U.S. CDC says there has been evidence of CHIKV transmission in Paraguay within the last 5 years.
During 2024, about 2.2 million people visited this South American country.
Furthermore, the CDC says certain travelers visiting Paraguay may be considered for Chikungunya vaccination. CHIKV vaccines are offered at travel clinics and pharmacies in the United States.
The São Paulo State Health Department recently confirmed that Dengue is a seasonal disease. During the rainy season, a high transmission rate can be observed. Throughout Brazil, Dengue is the most critical arbovirus disease transmitted by arthropod vectors.
Over the past 25 years, nearly 18 million Brazilians have been infected with Dengue.
As of May 26, 2025, the state of São Paulo, with a population of 44 million, has reported about 617,000 of Brazil's 2.5 million Dengue cases this year. According to data, the state of São Paulo leads in the number of deaths from dengue fever, with 692 cases.
In February 2025, a public health emergency was declared to alert residents and international visitors of this continuing health risk.
Last year, about 2.1 million Dengue cases and 2,100 related fatalities were reported in São Paulo.
This ongoing surge is related to Dengue virus serotype 3 (DENV-3) reemergence since the population is not immunized against this serotype.
While a second-generation Dengue vaccine will be limited in availability in 2025, Butantan Institute in São Paulo has been involved in developing an innovative vaccine that could become available in 2026.
Additionally, with the annual influenza season arriving in South America, beginning on May 27, 2025, the flu vaccination will be offered to the entire population in São Paulo's 645 municipalities over six months of age.
When visiting Brazil in 2025, the U.S. CDC recommends international travelers speak with a travel vaccine expert about various immunization options, such as chikungunya and yellow fever.
