Search API
With a population of 2.8 million, Jamaica remains a popular tourist destination in June 2025. Last year, this Caribbean Island destination welcomed over 4 million visitors by air and sea.
A short two-hour flight from Miami, Florida, brings tourists to vast resorts and warm waters.
While the U.S. Department of State recently reduced its Level 3 travel advisory for Jamaica, it still advises Americans to exercise caution while visiting in June 2025.
As of May 29, 2025, the State Department's periodic review indicates that civil unrest in Jamaica has decreased since 2024; however, it remains statistically high throughout the country.
Tourist areas typically experience lower rates of crime compared to other parts of the country. Still, the homicide rate reported by the Government of Jamaica is among the highest in the Western Hemisphere.
From a local health perspective, U.S. citizens should not expect the same level of healthcare services in Jamaica. This concern includes slower emergency service response times and reduced availability of care for illnesses or injuries.
Private hospitals typically require payment upfront before admitting patients and may not have the necessary resources to provide specialized care.
The U.S. Embassy in Jamaica previously stated 'Be aware that U.S. Medicare/Medicaid does not apply overseas. Most hospitals and doctors overseas do not accept U.S. health insurance. U.S. citizens with medical emergencies can face bills in the tens of thousands of dollars, with air ambulance service to the United States.'
We highly recommend that you purchase insurance before traveling, writes the Embassy, which is located at 142 Old Hope Road, Kingston 6.
Seperately, the U.S. CDC advises visitors to take actions to protect themselves from diseases such as measles and chikungunya.
Furthermore, the Pan American Health Organization has been working since 2003 to control and prevent dengue. The disease remains a substantial concern throughout the Americas, but only 165 cases have been reported in Jamaica this year.
The CDC recommends checking the vaccines and medicines list and visiting your doctor at least a month before your trip to Jamaica.
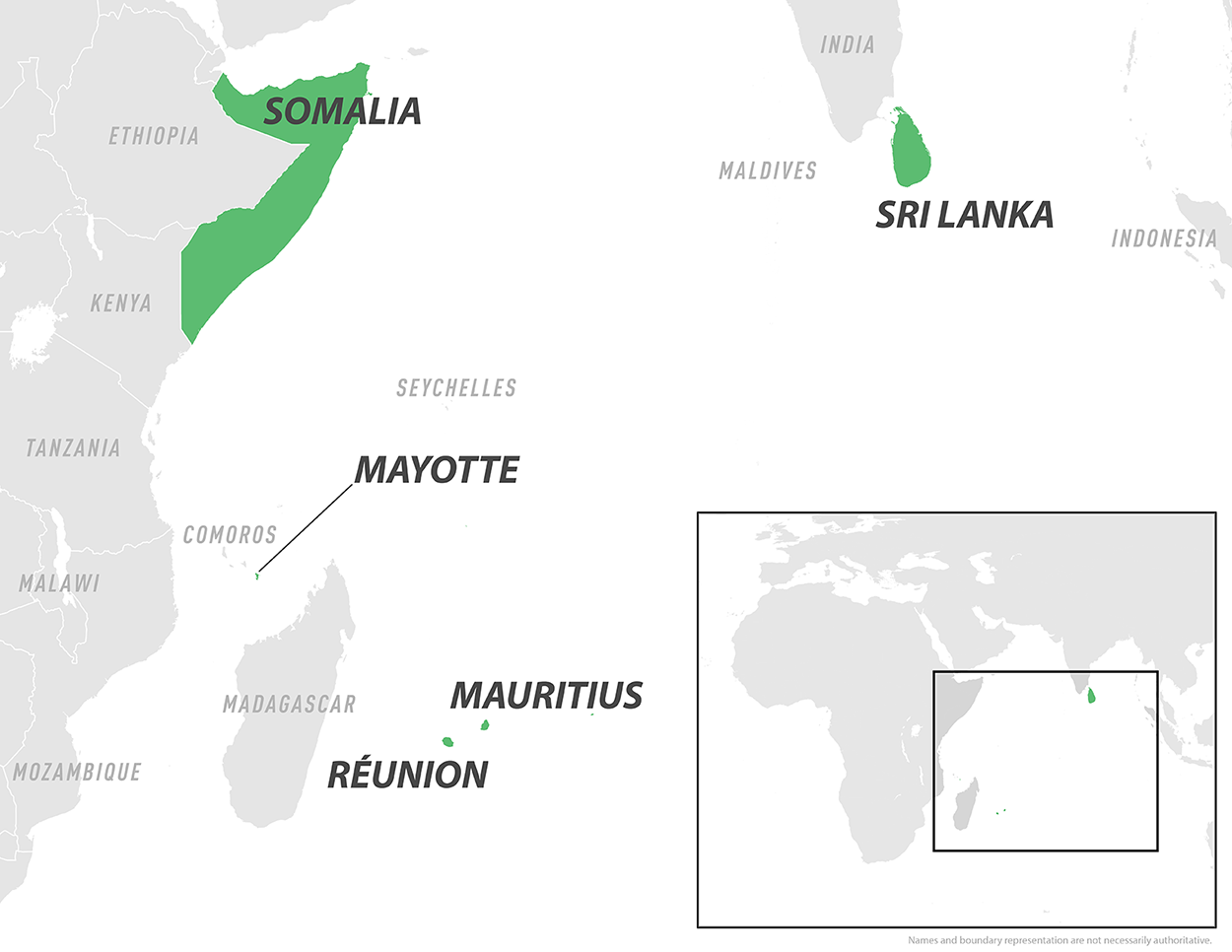
Since late May 2025, France's overseas department of Mayotte has experienced a significant outbreak of the mosquito-transmitted Chikungunya virus throughout the island.
As of June 2, 2025, Public Health France reports a total of 560 confirmed cases of chikungunya recorded during this phase of the ORSEC arbovirus plan.
From a location perspective, there has been a persistent concentration of cases in the municipalities of Mamoudzou, Pamandzi, and Dzaoudzi.
These are Mayotte's first locally transmitted cases of chikungunya since the 2005–2006 outbreak, which resulted in approximately 7,300 cases.
According to the World Health Organization (WHO), Chikungunya outbreaks have been documented on islands within and countries bordering the Indian and Pacific Oceans, such as Mauritius, Réunion, Somalia, and Sri Lanka, in 2025.
Public health response measures have included targeted vaccination efforts with Valneva SE's IXCHIQ® vaccine, the first approved monovalent, single-dose, live-attenuated vaccine.
While the WHO states that no measures related to international traffic and trade are warranted at this time, the U.S. CDC has updated its information to a Level 2 - Practice Enhanced Precautions, Travel Health Advisory.
The CDC recommends vaccination for travelers visiting an area with a chikungunya outbreak. Furthermore, if you are pregnant, consider reconsidering travel to the affected areas, particularly if you are nearing the delivery of your baby. Mothers infected around the time of delivery can pass the virus to their baby before or during delivery.
In addition to chikungunya, another mosquito-transmitted disease has been spreading on Mayotte. The circulation of dengue fever on the island has reached 21 confirmed cases since the beginning of 2025.
Before visiting Mayotte in June 2025, the CDC recommends consulting with a travel vaccine specialist to discuss immunization options.
ImmunityBio, Inc. recently announced that the U.S. Food and Drug Administration (FDA) has granted Expanded Access authorization for the use of its Cancer BioShield™ platform, anchored by ANKTIVA®, to treat lymphopenia in adult patients with refractory or relapsed solid tumors independent of tumor type who have progressed after first-line standard-of-care treatment, chemotherapy, radiation, or immunotherapy.
On June 2, 2025, ImmunityBio stated 'while oncologists and patients have long had therapies such as EPOGEN® and NEUPOGEN® to manage chemotherapy- and radiation-induced anemia and neutropenia, no comparable option has been available for lymphopenia.'
To date no treatment exists for lymphopenia, a depletion of critical lymphocytes responsible for immunogenic cell death, specifically natural killer (NK) cells, killer CD8+ T cells and CD4+ with memory T cells.
Treatment-induced lymphopenia is a debilitating consequence of chemotherapy, radiation, specific immunotherapies, and steroids. This treatment-acquired immunodeficiency not only increases susceptibility to infections but also deprives the body’s immune system to fight residual or recurrent cancer, accelerating metastasis and disease progression, and contributing to early mortality.
Countless publications over the last two decades have reported lymphopenia as a highly predictive biomarker of poor prognosis across all tumor types.1-6 Despite its significant clinical impact, the pharmaceutical industry has largely overlooked lymphopenia as a disease in its own right, and no approved therapies have existed to directly address it, until the approval of ANKTIVA in the treatment of BCG-unresponsive bladder cancer with the mechanism of action of an IL-15 superagonist proliferating key lymphocytes.
Dr. Patrick Soon-Shiong, Founder, Executive Chairman, and Global Chief Scientific and Medical Officer of ImmunityBio, commented in a related press release, “This FDA authorization allows all patients with solid tumors suffering from immune collapse following first-line therapy of chemo, radiation, or immunotherapy to access ANKTIVA."
"The survival benefit we observed at ASCO 2025 in 3rd to 6th line advanced metastatic pancreatic cancer confirms that restoring lymphocyte levels—rather than depleting them—can change the course of disease.”
As of June 4, 2025, ANKTIVA is available at participating cancer centers in the United States and other countries, such as the Kingdom of Saudi Arabia.

With measles cases being reported in many countries this year, the U.S. Centers for Disease Control and Prevention (CDC) issued a Travel Health Advisory highlighting updated vaccination guidance regarding this highly contagious respiratory illness.
As of May 28, 2025, the CDC states that international travelers are at risk of contracting measles if they have not been fully vaccinated at least two weeks before departure or have not had measles in the past.
Therefore, the CDC recommends that 'all international travelers should be fully vaccinated against measles with the measles-mumps-rubella (MMR) vaccine, according to the CDC's measles vaccination recommendations for international travel.
This is essential guidance since most people who bring measles into the United States are unvaccinated U.S. residents who get infected during international travel.
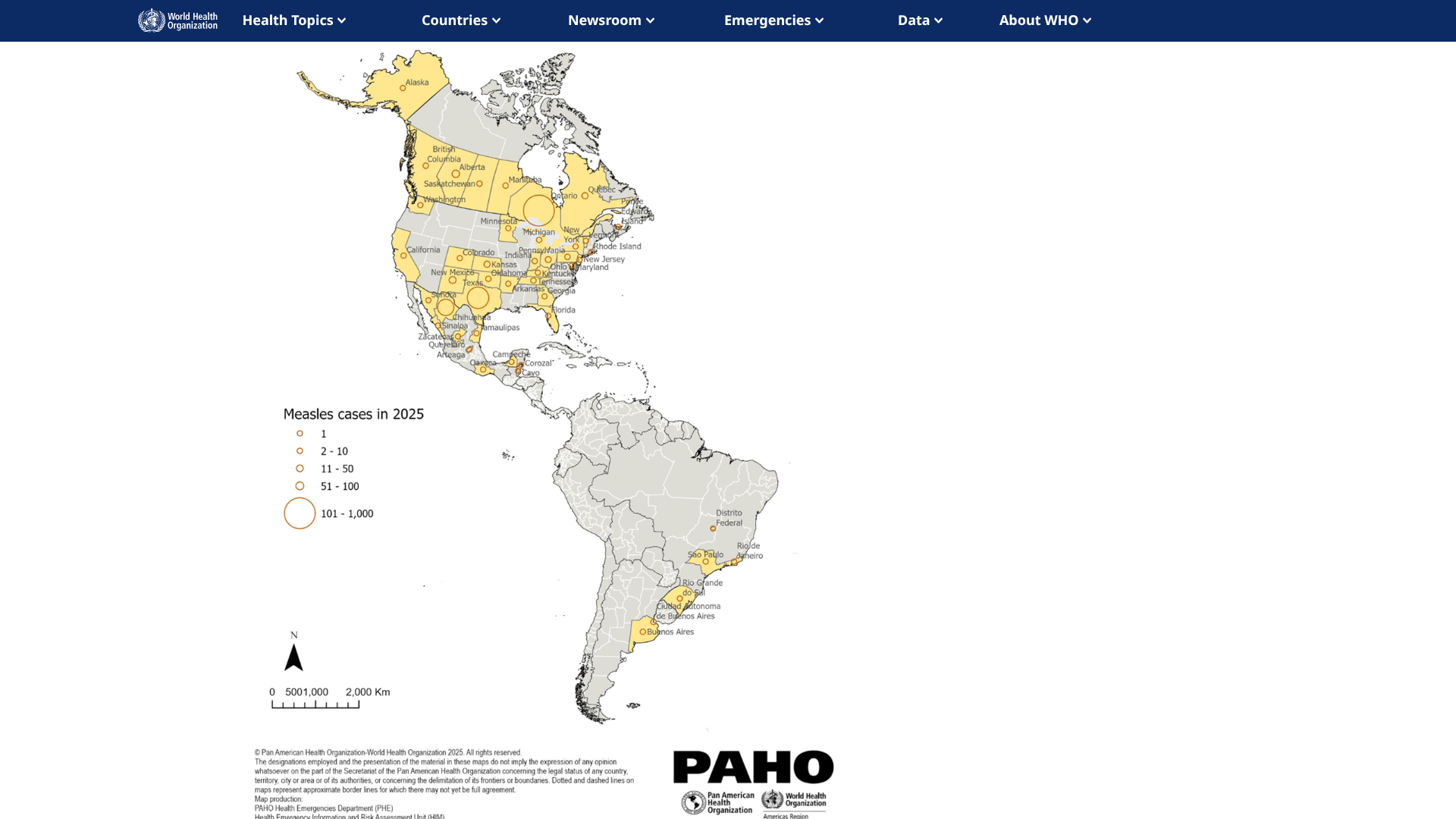
Currently, significant measles outbreaks have been confirmed in the Region of the Americas, including Canada (2,755 cases), Mexico, and Texas.
While the Texas measles outbreak has significantly slowed, the outbreak in Ottawa, Canada, continues to expand, with 1,949 cases reported.
As of June 4, 2025, various measles-containing vaccines are available at health clinics and pharmacies in the United States.
The Republic of Ecuador has been exposed to a wide range of zoonotic pathogens that have a significant impact on the population's health and the overall economy.
Recent disease outbreaks, such as yellow fever, rabies, hantavirus, and highly pathogenic avian influenza A(H5N1), have disrupted the health system of this South American country.
For example, the number of yellow fever cases reported in South America so far in 2025 represents a threefold increase compared to the cases reported in 2024.
Since the beginning of 2025 and as of early May, four confirmed fatal cases of yellow fever have been reported in Ecuador, from the provinces of Morona Santiago (one fatal case) and Zamora Chinchipe (three fatal cases).
These conditions led Ecuador to establish priority surveillance, prevention, and control strategies for potential outbreaks of animal-borne diseases.
In response to these conditions, with technical support from the Pan American Health Organization/World Health Organization, specialists from the Ecuador Ministry of Public Health, and others announced on June 2, 2025, they are working to prioritize zoonotic and emerging diseases to obtain a list that would provide the country with clarity in the implementation of surveillance and control tasks for these pathologies.
To alert international visitors, the U.S. CDC has included Ecuador in various disease outbreak alerts.
The CDC recommends yellow fever vaccination for travelers aged 9 months or older traveling to areas below 7,550 feet in elevation, east of the Andes Mountains, in the provinces of Morona-Santiago, Napo, Orellana, Pastaza, Sucumbíos, Tungurahua, and Zamora-Chinchipe.
As of June 3, 2025, the YF-Vax vaccine is generally not recommended for travel to areas with an elevation below 7,550 ft.
The vaccine is required for most travelers arriving from Brazil, the Democratic Republic of the Congo, or Uganda, including those with 12-hour airport transits or layovers in any of these countries. But not those arriving from the U.S.
Located on South America's Pacific Ocean, Ecuador also includes the Galápagos Province, about 600 miles west of the mainland.
This travel vaccine, along with others, is commercially offered at clinics and pharmacies in the U.S.

The Colorado Department of Public Health (CDPH) and Environment, along with El Paso County Public Health, have recently confirmed six measles cases that passed through Denver International Airport, which set an all-time passenger record in 2024, serving 82,358,744 passengers.
In May 2025, measles was confirmed in two unvaccinated residents of El Paso County. These adults are unrelated, but were at Denver around the same time on May 14, 2025.
Both individuals are recovering at home.
In addition, a third Colorado passenger on Turkish Airlines who arrived in Denver on May 13, 2025, has been confirmed positive for measles. The vaccinated adult from Arapahoe County is recovering at home. No public exposures have been identified in connection with this case.
According to a CDPH press release on June 1, 2025, this brings the total number of cases associated with an out-of-state traveler who flew while infectious to six: four passengers on the flight (three Colorado residents and one out-of-state resident) and two El Paso County residents who were at the airport during the exposure period.
Separately, the U.S. CDC published a Global Measles Outbreak Travel Health Advisory on May 28, 2025.
Measles is highly contagious and can sometimes lead to serious health problems, but it is a vaccine-preventable disease. The measles, mumps, and rubella (MMR) vaccine provides strong protection and is generally available at health clinics and pharmacies in the U.S.

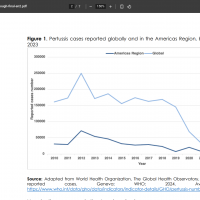
Since April 2024, the Ministry of Health (MOH) of the Republic of Costa Rica has reported an outbreak of Bordetella pertussis (whooping cough) in the Tibas district of San Jose, which has a population of over 70,000 residents.
As of June 2, 2025, the MOH remains on alert for whooping cough cases in local schools, which are primarily transmitted through respiratory droplets from coughing or sneezing, and have an incubation period ranging from 4 to 21 days.
On May 20, 2025, a case of whooping cough was confirmed in a teacher at a school in Golfito and a resident of the Corredores canton. There are now 14 confirmed cases in home isolation, in good general health, and receiving treatment and monitoring from the inter-institutional team.
Following the initial diagnosis, 79 direct contacts were identified, including 72 students, six teachers, and one cleaning staff member. Of these, 31 people presented symptoms.
The MOH recommends that the local population comply with the national vaccination schedule: the Pentavalent vaccine at 2, 4, 6, and 15 months, the Tetravalent vaccine at 4 years old, and the vaccine for pregnant women, indicated from the 20th week of pregnancy.
About 18 years ago, Costa Rico declared a whooping cough emergency and launched a nationwide vaccination program.
While the U.S. CDC advises individuals planning to visit Costa Rica in June 2025 to stay up to date with routine vaccinations, the agency also highlights the presence of Chikungunya and measles in Costa Rica.
These travel vaccines are commercially offered at clinics and pharmacies in the U.S. and should be administered about one month before departure abroad.
After six years without reporting a Zika virus case, the Hawaiʻi Department of Health (DOH) is investigating travel-related cases involving two individuals.
On May 27, 2025, the DOH announced that vector control teams are responding and will continue operations in areas where individuals have spent time, including the Waialua/Haleʻiwa area on the island of Oʻahu.
While Hawaiʻi has mosquito species capable of transmitting Zika, but the virus is not established in the state. As of June 2025, no locally acquired cases have been documented in Hawaiʻi.
The most recent travel-related Zika case in Hawaiʻi was reported in 2019. Travel-associated cases were more frequently reported in Hawai‘i during 2015-2019 when Zika was circulating globally, peaking at 25 cases in 2017.
Over the last few years, there has been a significant increase in Zika cases reported in the Americas, including in Puerto Rico. During 2025, over 12,600 cases have already been reported.
The DOH says Zika can also be transmitted through sex from a person who has Zika. The virus has been found in semen, vaginal fluids, saliva, urine and breast milk.
While most people recover from this mosquito-transmitted disease, pregnant women and their unborn children may become infected, leading to serious health issues.
A study published in January 2025 found that the mortality rates among 11.4 million children born with a severe Zika virus infection were higher. Young children with congenital Zika syndrome (CZS) had a 13-fold higher risk of death compared with those without CZS.
This study found a cause-specific mortality hazard ratio of 30.28.
As of June 2, 2025, the U.S. Food and Drug Administration has not approved a vaccine that prevents Zika.
The U.S. CDC says, regardless of where you live or visit, if mosquitoes are active in your area, wear long clothing and use insect repellent to reduce the risk of bites. Additionally, travelers returning from an area with a risk of Zika should take steps to prevent mosquito bites for three weeks after their return.

Since being discovered in Lyme, Connecticut, decades ago, Lyme disease has expanded west in the United States, causing significant health risks to people when outdoors.
For example, the Michigan Department of Health and Human Services (MDHHS) recently confirmed Lyme disease is now the most common tick-borne disease in Michigan, and anaplasmosis is increasing across the state.
Lyme disease cases in Michigan have increased by 168% over the last five years.
Michigan recorded 1,215 cases in 2024, as compared to 452 cases in 2020.
Anaplasmosis cases in Michigan have increased by almost fivefold over the last five years, with 82 cases reported in 2024 compared to 17 in 2020.
"Preventing tick bites is the best way to prevent tick-borne diseases, including Lyme disease and anaplasmosis," said Dr. Natasha Bagdasarian, MDHHS's chief medical executive, in a media release on May 21, 2025.
"If you find a tick attached to your body, promptly remove it. Monitor your health, and if you experience fever, rash, muscle or joint aches, or other symptoms, or if you suspect a tick has been attached for more than 24 hours, consult with your medical provider."
As of June 2, 2025, the U.S. Food and Drug Administration has not approved a vaccine for the prevention of Lyme disease. However, an innovative vaccine candidate (VLA15) is progressing in late-stage clinical research.
VLA15 is a multivalent recombinant protein vaccine targeting Borrelia's outer surface protein A (OspA). It is designed for prophylactic, active immunization against Lyme disease. The first data readout of the Phase 3 clinical trial is expected by the end of 2025.