Search API
Acne, a common skin condition, has long been dismissed as a rite of passage for millions of adolescents for years. The U.S. NIH says hormones and genetics play a role in acne cases, but it is primarily driven by inflammation and lesions caused by bacteria that grow in clogged pores.
Sebum, oil that helps keep skin from drying out, and dead skin cells plug the pores, which leads to outbreaks of lesions, commonly called pimples or zits.
Most often, outbreaks occur on the face but can also appear on the back, chest, and shoulders, as stated by the NIH.
But vaccine researchers are attempting to change how to manage this unfortunate issue..
According to an article written by Benjamin Plackett and published by Nature on August 27, 2025, two vaccine candidates that aim to engage the immune system in tackling the underlying cause of acne.
The vaccines, one that treats the condition and one that acts as a preventive measure, both attempt to provoke the immune system into targeting these acne-causing bacteria.
“It has taken time for the field to treat acne as a disease of immune regulation, rather than just a surface issue," says dermatologist Anjali Mahto, spokesperson for the London-based British Skin Foundation.
"What matters most is this shift in mindset."
The complete, unedited article is posted at this link.

Numerous studies have indicated an increased risk of stroke and myocardial infarction (MI) following herpes zoster (HZ); however, the impact of vaccination remains uncertain, wrote researchers in a global analysis.
To assess the effectiveness of HZ (shingles) vaccination with recombinant zoster vaccine (RZV) or zoster vaccine live-attenuated (ZVL) against cardiovascular (CV) events in adults, numerous phase 3 and observational studies were assessed.
Across these studies, any HZ vaccination (RZV or ZVL) was associated with a significantly lower risk of stroke and MI, versus no HZ vaccination.
The pooled RR of 0.82 (95% CI 0.76–0.87) in adults ≥18 years and RR of 0.84 (0.82–0.87) in adults ≥50 years.
Thus, vaccine effectiveness was 18% (13–24%) and 16% (13–18%) in preventing CV events, respectively.
RZV vaccination was associated with a significantly lower risk of stroke and MI versus no HZ vaccination: pooled RR 0.79 (0.65–0.97) in adults ≥18 years and RR 0.79 (0.64–0.97) in adults ≥50 years, with a vaccine effectiveness of 21% (3–35%) and 21% (3–36%), respectively.
These researchers concluded that HZ vaccination was associated with a significantly lower rate of cardiovascular events.
Study author Dr. Charles Williams, Global Associate Medical Director for Vaccines at GSK, commented in a media release, "Further research studies are now needed to find out whether this association can be attributed to an effect of herpes zoster vaccination."
This assessment is scheduled to be presented on August 30, 2025.
As of August 29, 2025, shingles vaccination services are offered at most pharmacies and clinics in the United States and the United Kingdom.

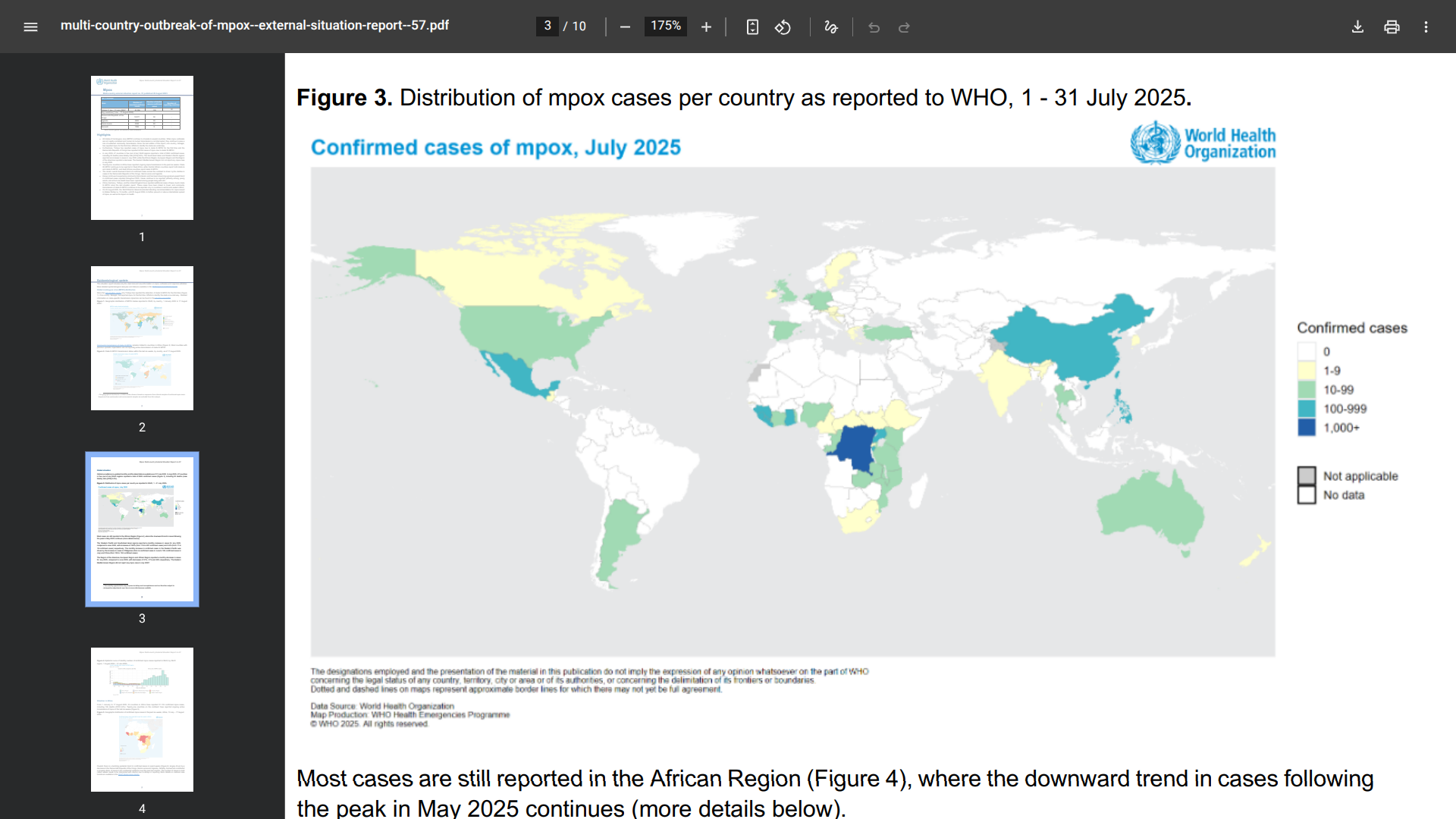
The WHO today published its Multi-country outbreak of mpox External situation report #57.
The World Health Organization (WHO) stated on August 28, 2025, that all monkeypox virus (MPXV) clades continue to circulate in several countries. When mpox outbreaks are not rapidly contained and human-to-human transmission is not interrupted, they continue to pose a risk of sustained community transmission.
In July 2025, 47 countries in five (out of six) WHO regions reported a total of 3,924 confirmed cases, including 30 deaths (case fatality ratio 0.8%).
The South-East Asian and Western Pacific regions reported an increase in cases in July 2025, while the African Region, European Region, and the Region of the Americas reported a decrease.
To alert international travelers to this serious health risk, the WHO Director-General has extended the standing recommendations for mpox issued to States Parties until August 20, 2026, to prevent further spread or reduce the international spread of mpox, as well as its impact on health.
The WHO and the U.S. CDC recommend mpox vaccination for those with the highest risk profile.
In June, the Institut Pasteur confirmed an unprecedented situation this summer, with locally acquired, mosquito-transmitted disease accelerating in France.
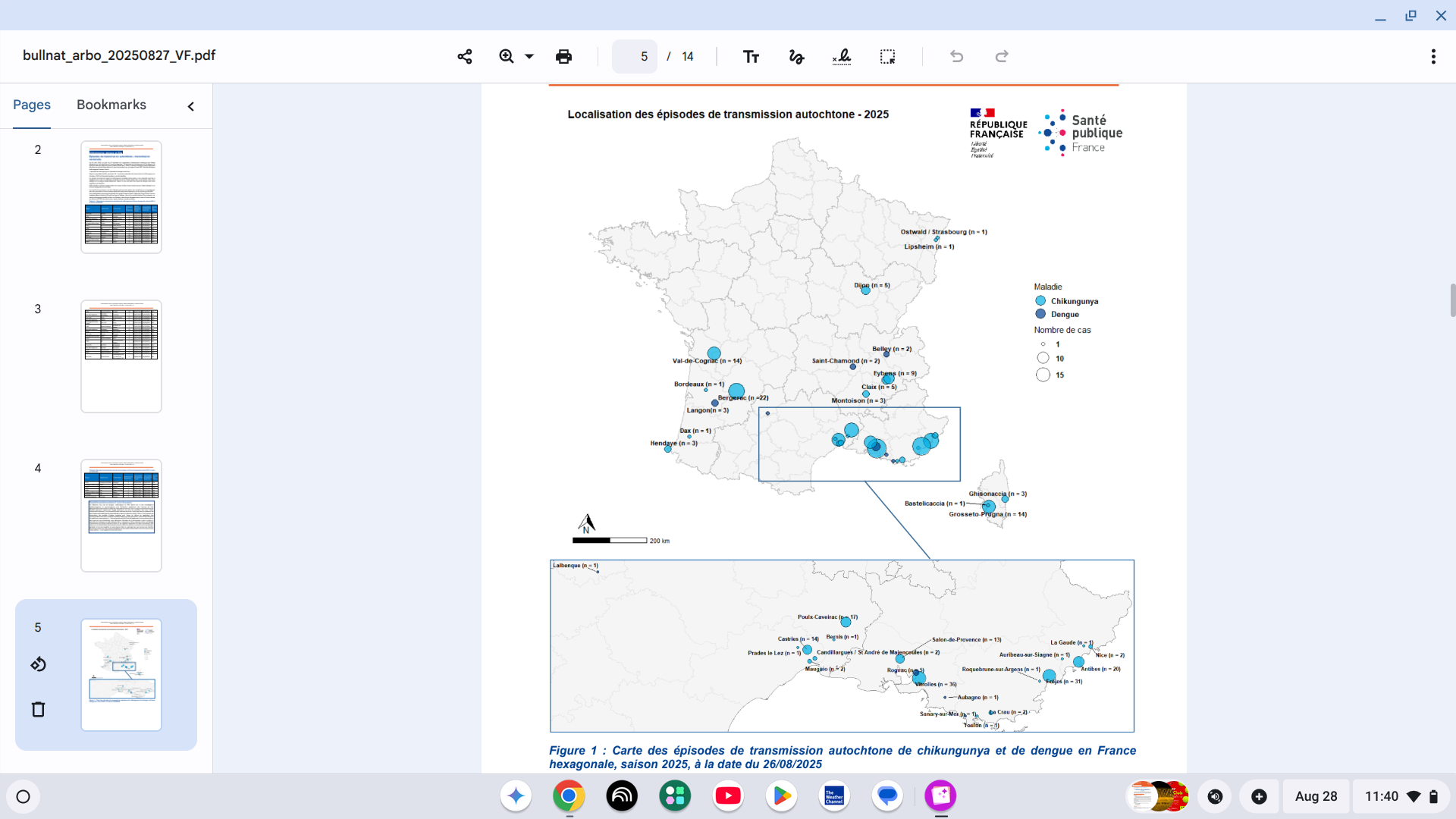
As of August 26, 2025, Public Health France reported 37 episodes of indigenous vector transmission have been identified in mainland France:
There have been 30 episodes of chikungunya, resulting in a total of 228 cases.
And seven separate episodes of dengue fever, totaling 15 cases.
They are located in the Provence-Alpes-Côte d'Azur, Corsica, Occitanie, Auvergne-Rhône-Alpes regions, already affected in previous years, and for the first time in Grand Est, Nouvelle-Aquitaine, and Bourgogne-Franche-Comté.
Furthermore, 15 human cases of vector-borne West Nile virus infection have been identified in five departments of mainland France. The affected regions are PACA, Occitanie, and, for the first time, Île-de-France.
Additionally, travel-related cases continue to be confirmed in 2025.
France has reported 946 imported cases of chikungunya, 825 imported cases of dengue fever, and seven imported cases of Zika.
When visiting France this year, the U.S. CDC offers various travel vaccine recommendations for anyone visiting disease outbreak areas. Currently, there are no vaccines available for West Nile or Zika viruses.
However, chikungunya vaccination services are offered in both France and the USA.

In light of the resurgence of pertussis across several countries in the region and the emergence and spread of antibiotic-resistant strains, the Pan American Health Organization (PAHO) reiterated the importance of strengthening vaccination and surveillance systems.
Known as Whooping cough, pertussis, is a very contagious respiratory illness that has resurged in the Americas. While 4,139 cases were reported in 2023, the number soared to 43,751 in 2024.
In the United States, preliminary data show that more than six times as many cases were reported in 2024 compared to 2023.
In the first seven months of 2025, nine countries, including Brazil, Mexico, Peru, and the U.S., reported over 18,595 cases and 128 deaths.
The PAHO states that the resurgence is linked to declining vaccination rates and emphasizes the need for strengthened, standardized surveillance.
Regional coverage for the first and third vaccine doses dropped to historic lows of 87% and 81% in 2021, respectively.
By 2023, a partial recovery was observed (90% and 88%), but these rates remain below the 95% recommended by PAHO, with significant disparities within countries.
Pilar Ramón-Pardo, head of PAHO's Special Program on Antimicrobial Resistance, commented in a media release on August 26, 2025, "We still have time to contain this issue, but we must act now: increase vaccination coverage, strengthen early detection, and enhance our outbreak response capacity."
When visiting countries such as Brazil, the U.S. CDC recommends travelers be protected against pertussis. Specifically, vaccination is essential from pregnant women and infants visiting outbreak areas.
Various pertussis vaccines are offered at clinics and pharmacies in August 2025.

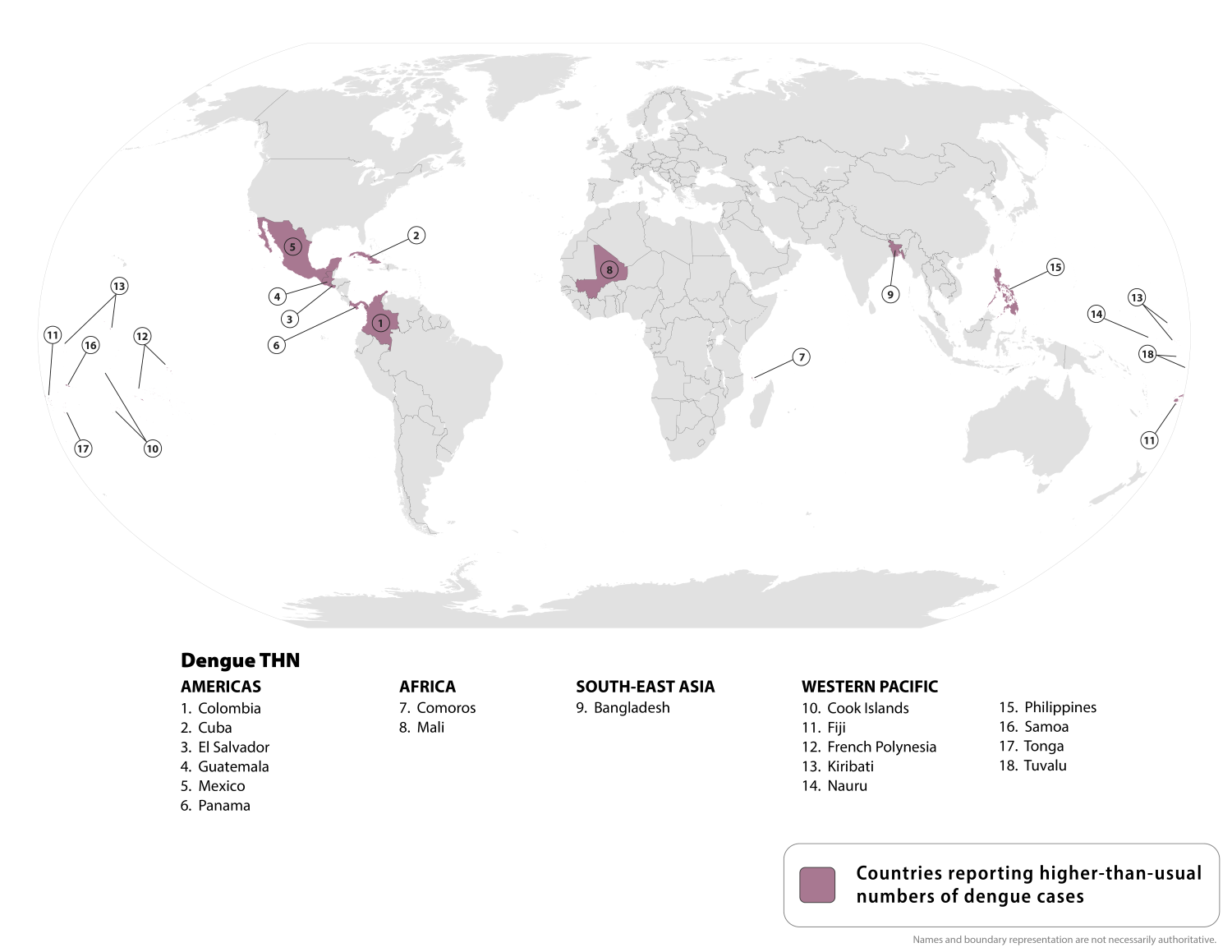
While the U.S. Centers for Disease Control and Prevention (CDC) states that Dengue is a year-round risk in many parts of the world, some countries are reporting an increase in cases of this mosquito-borne disease this year.
Since the beginning of 2025, over 4 million Dengue cases and over 2,500 dengue-related deaths have been reported from 101 countries/territories.
On August 21, 2025, the CDC updated its Travel Health Advisory, identifying a higher-than-expected number of dengue cases among U.S. travelers returning from these 18 countries.
Should you visit these areas, the CDC says to prevent mosquito bites, use an EPA-registered insect repellent, wear long-sleeved shirts and long pants when outdoors, and sleep in an air-conditioned room or a room with window screens.
The disease can take up to two weeks to develop, with illness generally lasting less than a week. Symptoms of Dengue include fever, headache, nausea, vomiting, rash, muscle and joint pain, and minor bleeding.
Currently, U.S. FDA-approved Dengue vaccines are only available in Puerto Rico, where Dengue has become endemic.
The second-generation vaccination is available in various countries, but not in the USA.