Search API
French Polynesia, located in the southwest Pacific Ocean, has been a dream vacation destination for decades.
However, these islands are also home to dengue fever, which is transmitted by the bite of infected mosquitoes.
According to Bulletin de surveillance sanitaire de Polynésie française n°34, published on September 2, 2025, there were 22 new dengue cases (19 confirmed last week.
And the blue alert for DENV-1 remains in effect.
Since November 2023, a total of 2,577 cases have been recorded.
To alert international travelers of this health risk, the U.S. CDC's Travel Health Advisory issued on August 21, 2025, included French Polynesia.
The CDC identified the island groups of the Society Islands (Tahiti, Moorea, and Bora Bora), the Marquesas Islands (Hiva Oa and Ua Huka), and the Austral Islands (Tubuai and Rurutu).
The CDC reported that these countries are experiencing higher-than-usual numbers of cases and have identified a higher-than-expected number of dengue cases among U.S. travelers returning from those countries.
While a second-generation dengue vaccine is available in various countries, it is not offered in the USA.
Fortunately, several dengue vaccine candidates are conducting late-stage research focused on preventing all four virus types from infecting people.

Mumbai, the capital city of the Indian state of Maharashtra, with a population of over 12 million residents, recently reported a significant increase in vector-borne diseases in 2025.
As of September 2025, the Brihanmumbai Municipal Corporation's (BMC) health report reveals a surge in cases of chikungunya, dengue, and malaria, which are impacting Mumbai's residents, formerly known as Bombay.
Daijiworld Media reported BMC data shows Mumbai has confirmed 5,706 malaria cases, a 42% rise compared to 2024.
Chikungunya cases increased from 210 in 2024 to 485 this year.
Dengue cases increased from approximately 1,979 to 2,319 in 2025.
To contain the spread, the BMC has restarted its 'Zero Mosquito Breeding' initiative to reduce the number of virus-carrying vectors.
BMC officials said cases are expected to decline once monsoon activity eases.
To alert international visitors, both the U.S. CDC and the UK government have issued travel vaccine advice for those planning trips to Maharashtra in 2025. Mumbai attracts almost 6 million tourists annually.
The CDC recommends that chikungunya vaccination may be considered for individuals traveling to or residing in this location for an extended period (e.g., 6 months or more).

The World Health Organization (WHO) recently confirmed that the global cholera situation continues to deteriorate in 2025.
Cholera is resurging in several countries, with some that had not reported substantial case numbers in years.
Since January and through August 17, 2025, the WHO has reported a total of 4,738 cholera/Acute Watery Diarrhoea-related fatalities from 31 countries, with six of the 31 countries reporting case fatality rates (CFR) above 1%.
For example, suspected cholera cases have been reported from two provinces of the Republic of Chad. Among these, Chokoyane is the most affected area, accounting for 541 cases and 25 deaths, with a district-specific CFR of 4.6%.
While Cholera is an acute diarrheal infection caused by consuming food or water contaminated with the bacterium Vibrio cholerae, primarily associated with poor sanitation and limited access to safe water, it can be prevented with vaccination.
As of September 5, 2025, the U.S. CDC's Travel Health Advisories for these countries advise cholera vaccination before departing abroad. Most cholera cases diagnosed in the U.S. are related to international travel.
In the U.S., Vaxchora® (CVD 103-HgR) is the only U.S. FDA-approved vaccine available at travel clinics and pharmacies.


Chikungunya virus disease patients have been reported in 14 countries/territories in Europe this year. Several of these countries are positioned along the Mediterranean Sea.
In mainland France, the Health Agency reported on September 4, 2025, that an unprecedented situation has developed, with the number of locally acquired cases exceeding previously unseen levels.
As of early September 2025, 34 episodes of chikungunya had been reported, totaling 301 cases.
Mosquitos have transmitted the virus to people in the Provence-Alpes-Côte d'Azur, Corsica, Occitanie, Auvergne-Rhône-Alpes regions, already affected in previous years, and for the first time this year in Grand Est, Nouvelle-Aquitaine, and Bourgogne-Franche-Comté.
Additionally, 957 international travelers have been diagnosed with chikungunya while in France.
When planning a trip to France this fall, health agencies in the United Kingdom and the United States recommend speaking with a travel vaccine expert regarding chikungunya vaccination options.
In the U.S., travel clinics and pharmacies offer a U.S. FDA-approved chikungunya vaccine.
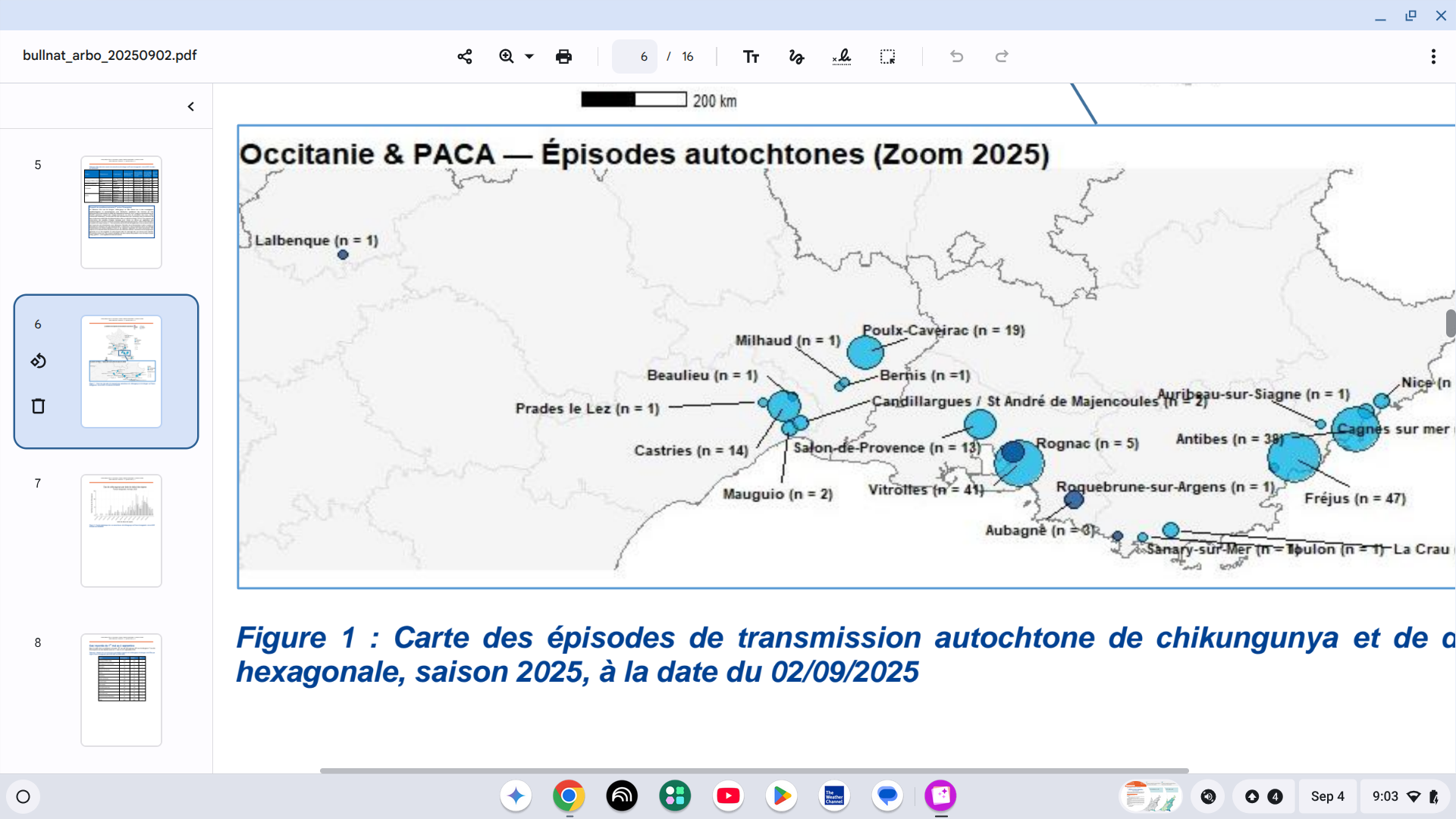

The U.S. Centers for Disease Control and Prevention (CDC) today announced an update to its Level 2 Travel Health Advisory for the Republic of Colombia, due to a yellow fever outbreak.
On September 3, 2025, the CDC reported an increase in cases in new areas in the South American country.
In a Facebook post, the Health Ministry reported that between 2024 and 2025, 132 yellow fever (YF) cases were registered, resulting in 57 deaths, in the following Colombian departments: Tolima, Huila, Cauca, Nariño, Putumayo, Caldas, Meta, Vaupés, Guaviare, and Caquetá.
The CDC recommends that travelers to these newly affected areas get vaccinated at least 10 days before traveling abroad, as yellow fever is a severe viral disease.
Last year, the Colombian government declared a nationwide health emergency to alert residents and visitors to the situation.
Colombia welcomed over 6.5 million international visitors.
Recently, the U.S. Embassy in Bogota issued an alert stating visitors should reconsider travel to the Valle del Cauca Department due to civil unrest.
As of September 2025, YF is a vaccine-preventable disease, and proof of vaccination is required to enter various countries, including Colombia.
A booster dose may be given to eligible travelers or those who received their last dose of the YF vaccine at least 10 years prior and will be in a higher-risk setting, according to the CDC.
"With yellow fever cases rising in Colombia, the CDC has expanded the list of areas where vaccination is now recommended. We're seeing a similar trend in parts of Peru and Bolivia, too," commented Jeri Beales, MSN, RN, BS.
This isn't a mosquito-borne virus to take lightly—yellow fever can be deadly, with fatality rates as high as 60% in severe cases."
"If you're planning a trip to Colombia, consult with a travel health provider about getting vaccinated. It's highly effective and usually offers lifelong protection. Still, it's only available at certified clinics, so schedule ahead—ideally a few weeks before you leave to give the vaccine time to create a robust immune response so you're fully protected before arriving," added Beales, vaccination leader at Destination Health, located in the greater Boston, MA, area.
In the United States, the YF-VAX vaccine is commercailly offered at travel vaccination retailers.
Note - vaccine expert insight added on September 6, 2025.
With Lyme disease cases steadily declining in the United States, the only vaccine candidate conducting late-stage clinical research announced very encouraging news today.
On September 3, 2025, Valneva SE reported positive immunogenicity and safety data from the ongoing Phase 2 study of Lyme disease vaccine candidate, VLA15.
The strong anamnestic immune response and favorable safety profile following a third booster dose were consistent with those reported after receiving previous annual booster doses, further demonstrating compatibility with the anticipated benefits of a yearly vaccination before each Lyme season, wrote the company.
Juan Carlos Jaramillo, M.D., Chief Medical Officer of Valneva, commented in a press release, "These latest data further reinforce the potential benefits of booster doses across all evaluated age groups.... as the disease continues to expand geographically, it remains a pressing unmet medical need affecting communities across the Northern Hemisphere."
"Each set of positive results moves us closer to the possibility of making this vaccine available to both adults and children living in Lyme-endemic areas."
Lyme disease (Lyme borreliosis) is a bacterial disease transmitted to humans through the bite of infected ticks, initially detected in 1977 in Lyme, Connecticut, and now found in most northeastern states.
In the UK, ticks that carry Lyme disease are most active in the spring and summer. Approximately 4% of ticks in England and Wales are infected with Lyme disease.
As of September 2025, the U.S. Centers for Disease Control and Prevention's Tick Bite Data Tracker displays case data and maps for the U.S.
The Communicable Diseases Agency of the Republic of Singapore has reported a total of 21 chikungunya cases this year.
As of August 28, 2025, this number has already surpassed the total recorded for all of 2024, which was 15 cases.
The increase began in May 2025, primarily due to travelers returning from areas affected by the chikungunya outbreak in China's Guangdong Province and several French Territories in the western Indian Ocean.
In 2024, this sovereign island country and city-state in Southeast Asia welcomed approximately 16.5 million international tourists, representing a 20% increase from the previous year.
In early August, Professor Ooi Eng Eong from Duke-NUS Medical School explained to the media that although chikungunya is less deadly than dengue, it can be debilitating. Chronic joint discomfort can limit physical activity and impact overall quality of life.
Although the U.S. CDC has not issued a Travel Health Advisory for chikungunya related to Singapore, the UK's Travel Health Pro indicates that there is a risk of chikungunya in this country.
As of September 3, 2025, the UK recommends that international travelers take precautions to avoid mosquito bites, especially during daytime hours.
Additionally, vaccination may be considered for individuals aged 12 years and older who meet specific eligibility criteria.
In the United States, the FDA has approved a chikungunya vaccine, which is available at travel clinics and pharmacies.
