Search API
The Brant County Health Unit (BCHU) has received confirmation of a human case of rabies in a resident of Brantford-Brant, Ontario, Canada.
As of September 6, 2024, the individual was hospitalized. As a precaution, family members and other close contacts are being assessed and offered post-exposure prophylaxis, as needed.
While the suspected exposure of the case was from a bat in the Gowganda area of the Timiskaming region, bats in all areas of Ontario are known to carry rabies. Bats, skunks, foxes, and raccoons are the most common to spread the rabies virus in Canada.
Ontario’s last domestic case of human rabies occurred in 1967, and there have been 26 human cases in Canada since 1924.
The World Health Organization (WHO) states that dogs spread most rabies cases globally. To date, there has never been a documented case of human-to-human transmission of rabies virus.
Once the virus infects the central nervous system and clinical symptoms appear, rabies is fatal in 100% of cases. There are an estimated 59,000 deaths from rabies annually. However, due to underreporting, documented case numbers often differ from the estimate, says the WHO.
Even though rabies is well-controlled in the United States, over 4 million Americans report animal bites each year, with 800,000 seeking medical attention.
In 2022, 54 U.S. jurisdictions reported 3,579 animal rabies cases. However, only ten people die from rabies infection annually in the U.S.
Rabies is a viral infection that causes brain and spinal cord inflammation. It is typically spread to humans through direct contact with saliva or mucous of an infected animal, such as through a bite or scratch. Even tiny bites or scratches, which can be challenging to see, can transmit the virus.
Effective rabies vaccines are available to immunize people before and after potential exposures.
As of 2024, three WHO pre-qualified human rabies vaccines, RABIVAX-S, VaxiRab N, and VERORAB, are available globally. In the United States, rabies vaccines are generally offered at travel clinics and pharmacies.
Note: This news article was updated on Sept. 28, 2024.
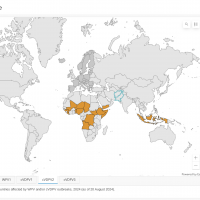
According to this week's Global Polio Eradication Initiative (GPEI) report, various countries confirmed poliovirus detections and vaccination efforts.
As of early September 2024, a two-round polio vaccination campaign was launched in the Palestinian territory, providing more than 640,000 children with two drops of novel oral polio vaccine type 2 (nOPV2) vaccine.
Additionally, new polioviruses cases and positive environmental isolates were detected in the following countries as of September 4, 2024:
Afghanistan: 1 WPV1 case
Pakistan: 8 WPV1-positive environmental samples
Côte d’Ivoire: 1 cVDPV2-positive environmental sample
DR Congo: two cVDPV2 cases
Egypt: 1 cVDPV2-positive environmental sample
Indonesia: 3 cVDPV2 cases
Niger: 2 cVDPV2 cases and 3 cVDPV2-positive environmental samples
Nigeria: 7 cVDPV2 cases and 1 cVDPV2-positive environmental sample
South Sudan: 2 cVDPV2-positive environmental samples
Sudan: 1 cVDPV2-positive environmental sample
To alert international travelers of their potential polio risk, the U.S. Centers for Disease Control and Prevention (CDC) reissued a Global Polio Alert - Level 2, Practice Enhanced Precautions Travel Health Notice on August 20, 2024, regarding polio outbreaks and poliovirus detections in 37 countries.
The CDC says that before traveling to any destination listed, adults who have previously completed the full, routine polio vaccine series may receive a single, lifetime booster dose of the polio vaccine.
Polio vaccination services are generally available to local clinics and community pharmacies in the U.S.
New research by modeling experts shows that vaccinating against Lassa Fever—a viral disease—would prevent millions of people from falling ill.
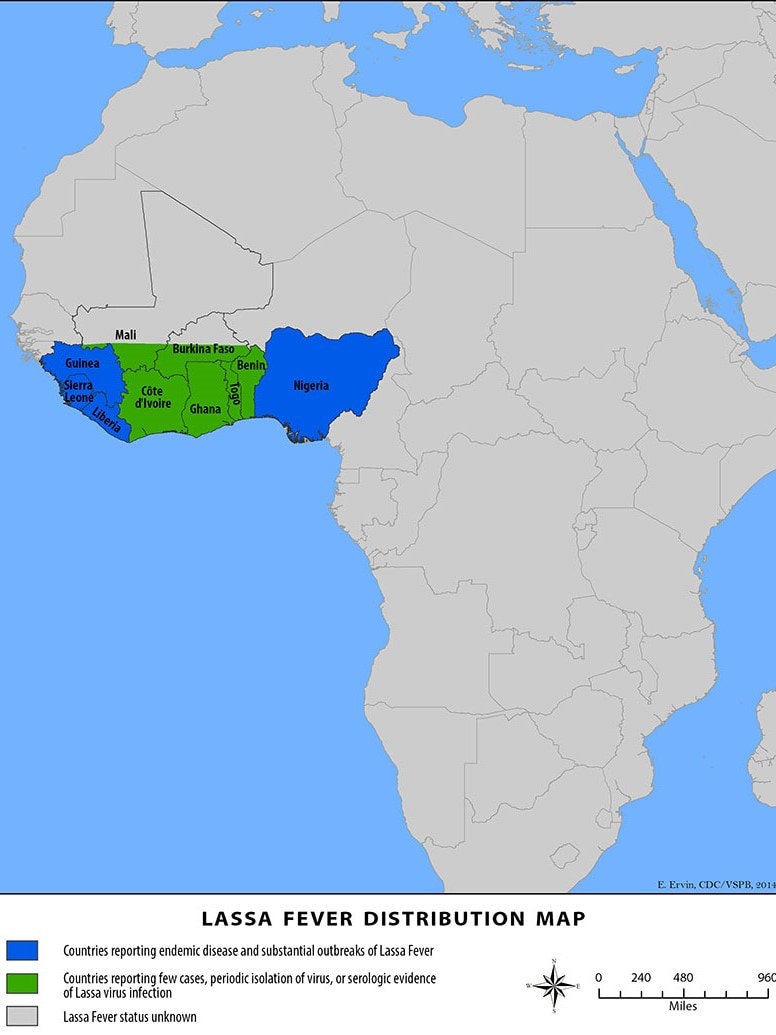
Lassa fever is found in parts of West Africa, including Sierra Leone, Liberia, Guinea, and Nigeria, where the first documented case occurred in 1969.
According to modeling by the Universities of Oxford and Liverpool and the Liverpool School of Tropical Medicine, deploying an effective Lassa vaccine across 15 countries of continental West Africa could save nearly 3,300 lives over ten years, wrote the Coalition for Epidemic Preparedness Innovations (CEPI).
The model was published on August 28, 2024, and predicted 2.7 million (95% uncertainty interval: 2.1–3.4 million) Lassa virus infections annually. However, due to limited access to diagnostics and healthcare, Lassa’s true disease burden could be much higher than reported.
They also model the emergence of ‘Lassa-X’—a hypothetical pandemic Lassa virus variant—and project impacts of achieving 100 Days Mission vaccination targets.
The results showed that around 5,500 lives could be saved and 33,000 hospitalizations avoided throughout a two-year outbreak if safe and 70% effective Lassa X vaccines were given to 40% of people per year starting within 100 days.
Overall, the most effective vaccination strategy was a population-wide preventive campaign targeting WHO-classified ‘endemic’ districts.
Richard Hatchett, CEO of CEPI, commented in a press release, “This study demonstrates the urgent need for a vaccine to protect people from this debilitating and sometimes deadly disease. Lassa fever has been a priority for CEPI since our launch in 2017, and we are proud to be one of the world’s leading Lassa vaccine R&D funders.”
CEPI is one of the world’s leading Lassa vaccine candidate R&D funders. To date, it has invested in six potential vaccine candidates, of which four have progressed into human testing.
One of CEPI’s partners, IAVI, has launched the first-ever Phase II clinical trial of a Lassa vaccine in Abuja, Nigeria.
In the northeastern section of the United States, concerns about the impact of Eastern equine encephalitis (EEE) and West Nile virus (WNV) have continued into September 2024.
The Massachusetts Department of Public Health (DPH) announced on September 5, 2024, one additional human case of EEE and one additional human case of WNV in Massachusetts this year.
The total number of EEE cases in Massachusetts this year is three, with seven WNV cases.
WNV risk levels in the following communities are being raised to high: Stoneham and Wakefield in Middlesex County.
Throughout the U.S., 38 states have reported WNV cases in 2024.
The last outbreak of EEE occurred in 2019-2020 and resulted in 17 human cases and seven deaths. In 2023, there were six human cases of WNV.
As of September, EEE risk levels have been raised to high in Acton, Ayer, Boxborough, Carlisle, Littleton in Middlesex County, and Harvard in Worcester County. The following communities are being raised to moderate: Bedford, Billerica, Chelmsford, Concord, Framingham, Groton, Lincoln, Shirley, Stow, Tyngsborough, Wayland, and Westford in Middlesex County; and Berlin, Bolton, Clinton, and Lancaster in Worcester County.
Public Health Commissioner Robbie Goldstein, MD, PhD., commented in a press release, “It is essential that residents continue to use mosquito repellent with an EPA-registered active ingredient every time they are outdoors. We also strongly recommend that residents and towns in high-risk areas for EEE reschedule their evening outdoor events to avoid peak mosquito biting hours.”
EEE and WNV are transmitted to humans through the bite of an infected mosquito.
“Mosquito behavior starts to change in September,” said State Epidemiologist Dr. Catherine Brown. “They will be less active during cooler temperatures. However, during warmer weather, such as being forecast for the end of next week, mosquitoes will be out and looking for their next meal.”
To the north of Massachusetts, the New Hampshire Department of Health and Human Services reported on September 4, 2024, that ten mosquito batches tested positive for EEE this year, with one fatal human case.
The last reported human cases of EEE in New Hampshire were in 2014 when three cases were identified. Two of those patients died.
As of September 6, 2024, no vaccines are available for people to protect themselves against EEE or WNV.
Novavax Inc. announced on X today that its updated 2024-2025 formula COVID-19 vaccine (NVX-CoV2705) has received Marketing Approval from Japan’s Ministry of Health, Labour and Welfare.
As of September 6, 2024, Novavax's vaccine is the only non-mRNA, protein-based COVID-19 vaccine available in the U.S. following the Food and Drug Administration (FDA) granting emergency use authorization.
John C. Jacobs, President, and Chief Executive Officer, Novavax, commented in a recent press release, "Our updated vaccine targets JN.1, the 'parent strain' of currently circulating variants, and has shown robust cross-reactivity against JN.1 lineage viruses, including KP.2.3, KP.3, KP.3.1.1 and LB.1."
Since August 2020, Novavax and Takeda Pharmaceutical Company Limited have partnered to develop, manufacture, and commercialize Novavax’s COVID‑19 vaccines.
Before visiting Japan, the U.S. CDC recommends travelers speak with a healthcare provider one month before departure about their vaccination options.
During the global dengue virus outbreak in 2024, Puerto Rico was the most impacted area in the United States. The U.S. CDC says that Dengue is endemic in Puerto Rico, cases are increasing, and it issued a Dengue travel advisory earlier in 2024.
As of week #32 in August 2024, Puerto Rico reported 2,704 dengue cases, with San Juan reporting over 950 cases.
The local health department's report for August 2024 indicates that 1,234 dengue-related hospitalizations and two dengue-related deaths occurred this year.
Last year, 1,242 dengue cases were reported in Puerto Rico.
Since 2019, the U.S. Food and Drug Administration (FDA) has approved the Dengvaxia vaccine for certain people with laboratory evidence of a previous dengue infection and living in areas where Dengue is endemic, such as Puerto Rico.
However, in late August 2024, Puerto Rico's Health Department issued a Notice of Discontinuation regarding access to Dengvaxia.
This announcement indicates that millions of visitors to Puerto Rico in 2024 will be unable to access a Dengue-preventive vaccine.
The FDA may approve various dengue vaccines in the coming months and once again become available throughout the U.S.
Additionally, diseases such as Chikungunya have been transmitted to people by the Aedes aeqypti mosquito.
The good news is that Valneva SE's IXCHIQ® single-dose chikungunya vaccine is available in the U.S. at travel clinics and pharmacies.

